
10.11: Nucleophilicity of Sulfur Compounds
- Page ID
- 211672
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Compounds incorporating a C–S–H functional group are named thiols or mercaptans. Despite their similarity, they are stronger acids and more powerful nucleophiles than alcohols. The IUPAC name of (CH3)3C–SH is 2-methyl-2-propanethiol, commonly called tert-butyl mercaptan.
Nucleophilicity of Sulfur Compounds
Sulfur analogs of alcohols are called thiols or mercaptans, and ether analogs are called sulfides. The chemical behavior of thiols and sulfides contrasts with that of alcohols and ethers in some important ways. Since hydrogen sulfide (H2S) is a much stronger acid than water (by more than ten million fold), we expect, and find, thiols to be stronger acids than equivalent alcohols and phenols. Thiolate conjugate bases are easily formed, and have proven to be excellent nucleophiles in SN2 reactions of alkyl halides and tosylates.
R–S(–) Na(+) + (CH3)2CH–Br  (CH3)2CH–S–R + Na(+) Br(–)
(CH3)2CH–S–R + Na(+) Br(–)
Although the basicity of ethers is roughly a hundred times greater than that of equivalent sulfides, the nucleophilicity of sulfur is much greater than that of oxygen, leading to a number of interesting and useful electrophilic substitutions of sulfur that are not normally observed for oxygen. Sulfides, for example, react with alkyl halides to give ternary sulfonium salts (equation # 1) in the same manner that 3º-amines are alkylated to quaternary ammonium salts. Although equivalent oxonium salts of ethers are known, they are only prepared under extreme conditions, and are exceptionally reactive. Remarkably, sulfoxides (equation # 2), sulfinate salts (# 3) and sulfite anion (# 4) also alkylate on sulfur, despite the partial negative formal charge on oxygen and partial positive charge on sulfur.
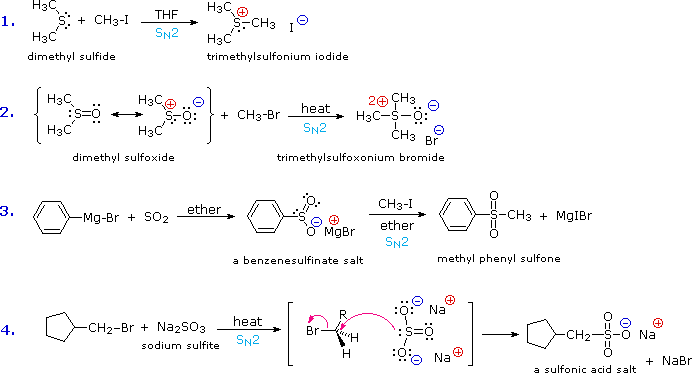
![]()
Oxidation States of Sulfur Compounds
Oxygen assumes only two oxidation states in its organic compounds (–1 in peroxides and –2 in other compounds). Sulfur, on the other hand, is found in oxidation states ranging from –2 to +6, as shown in the following table (some simple inorganic compounds are displayed in orange).
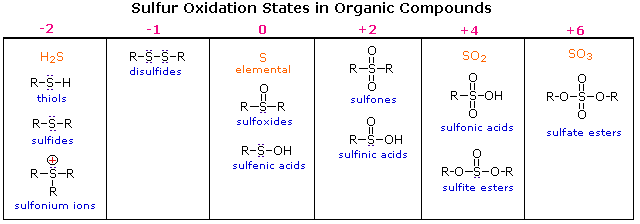
Try drawing Lewis-structures for the sulfur atoms in these compounds. If you restrict your formulas to valence shell electron octets, most of the higher oxidation states will have formal charge separation, as in equation 2 above. The formulas written here neutralize this charge separation by double bonding that expands the valence octet of sulfur. Indeed, the S=O double bonds do not consist of the customary σ & π-orbitals found in carbon double bonds. As a third row element, sulfur has five empty 3d-orbitals that may be used for p-d bonding in a fashion similar to p-p (π) bonding. In this way sulfur may expand an argon-like valence shell octet by two (e.g. sulfoxides) or four (e.g. sulfones) electrons. Sulfoxides have a fixed pyramidal shape (the sulfur non-bonding electron pair occupies one corner of a tetrahedron with sulfur at the center). Consequently, sulfoxides having two different alkyl or aryl substituents are chiral. Enantiomeric sulfoxides are stable and may be isolated.
Thiols also differ dramatically from alcohols in their oxidation chemistry. Oxidation of 1º and 2º-alcohols to aldehydes and ketones changes the oxidation state of carbon but not oxygen. Oxidation of thiols and other sulfur compounds changes the oxidation state of sulfur rather than carbon. We see some representative sulfur oxidations in the following examples. In the first case, mild oxidation converts thiols to disufides. An equivalent oxidation of alcohols to peroxides is not normally observed. The reasons for this different behavior are not hard to identify. The S–S single bond is nearly twice as strong as the O–O bond in peroxides, and the O–H bond is more than 25 kcal/mole stronger than an S–H bond. Thus, thermodynamics favors disulfide formation over peroxide.
Mild oxidation of disufides with chlorine gives alkylsulfenyl chlorides, but more vigorous oxidation forms sulfonic acids (2nd example). Finally, oxidation of sulfides with hydrogen peroxide (or peracids) leads first to sulfoxides and then to sulfones.
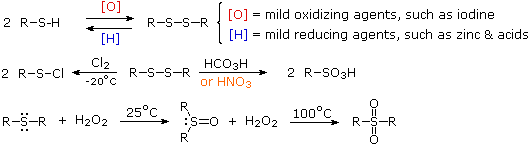
The nomenclature of sulfur compounds is generally straightforward. The prefix thio denotes replacement of a functional oxygen by sulfur. Thus, -SH is a thiol and C=S a thione. The prefix thia denotes replacement of a carbon atom in a chain or ring by sulfur, although a single ether-like sulfur is usually named as a sulfide. For example, C2H5SC3H7 is ethyl propyl sulfide and C2H5SCH2SC3H7 may be named 3,5-dithiaoctane. Sulfonates are sulfonate acid esters and sultones are the equivalent of lactones. Other names are noted in the table above.
Oxidation of Alcohols by DMSO
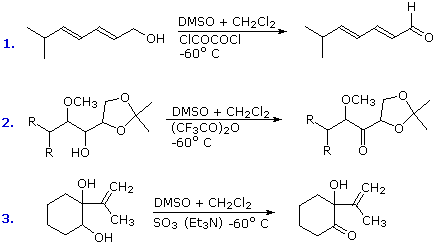
The conversion of 1º and 2º-alcohols to aldehydes and ketones is an important reaction which, in its simplest form, can be considered a dehydrogenation (loss of H2). By providing an oxygen source to fix the product hydrogen as water, the endothermic dehydrogenation process may be converted to a more favorable exothermic one. One source of oxygen that has proven effective for the oxidation of alcohols is the simple sulfoxide solvent, DMSO. The reaction is operationally easy: a DMSO solution of the alcohol is treated with one of several electrophilic dehydrating reagents (E). The alcohol is oxidized; DMSO is reduced to dimethyl sulfide; and water is taken up by the electrophile. Due to the exothermic nature of the reaction, it is usually run at -50 ºC or lower. Co-solvents such as methylene chloride or THF are needed, since pure DMSO freezes at 18º. The reaction of oxalyl chloride with DMSO may generate chlorodimethylsulfonium chloride which then oxidizes the alcohol (Swern Oxidation). Alternatively, a plausible general mechanism for this interesting and useful reaction is drawn below.
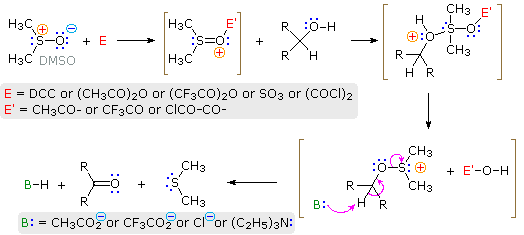
Because so many different electrophiles have been used to effect this oxidation, it is difficult to present a single general mechanism. Most of the electrophiles are good acylating reagents, so it is reasonable to expect an initial acylation of the sulfoxide oxygen. (The use of DCC as an acylation reagent was described elsewhere.) The electrophilic character of the sulfur atom is enhanced by acylation. Bonding of sulfur to the alcohol oxygen atom then follows. The remaining steps are eliminations, similar in nature to those proposed for other alcohol oxidations. In some cases triethyl amine is added to provide an additional base. Three examples of these DMSO oxidations are given in the following diagram. Note that this oxidation procedure is very mild and tolerates a variety of other functional groups, including those having oxidizable nitrogen and sulfur atoms.
Contributors
- William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry