
9.9: Carbocation Rearrangements
- Page ID
- 28203

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Hydride and Alkyl Shifts
Since the dehydration reaction of alcohol has a carbocation intermediate, hydride or alkyl shifts can occur which relocates the carbocation to a more stable position. The dehydrated products therefore are a mixture of alkenes, with and without carbocation rearrangement. Tertiary cation is more stable than secondary cation, which in turn is more stable than primary cation due to a phenomenon known as hyperconjugation, where the interaction between the filled orbitals of neighboring carbons and the singly occupied p orbital in the carbocation stabilizes the positive charge in carbocation.
- In hydride shifts, a secondary or tertiary hydrogen from a carbon next to the original carbocation takes both of its electrons to the cation site, swapping place with the carbocation and renders it a more stable secondary or tertiary cation.

Similarly, when there is no hydride available for hydride shifting, an alkyl group can take its bonding electrons and swap place with an adjacent cation, a process known as alkyl shift.
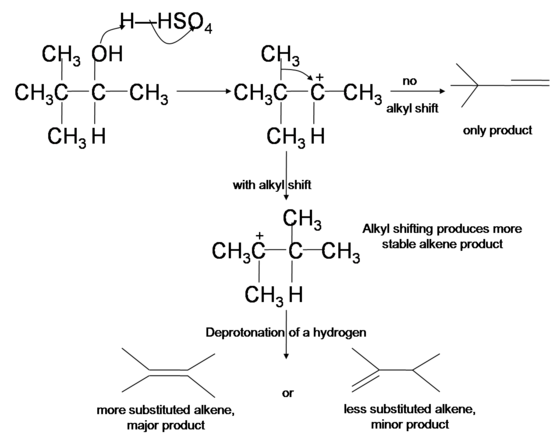
Consider the major product of the addition of HCl to 3,3-dimethyl-1-butene:

This may not be the result that you would have predicted, based on what we have learned so far about electrophilic additions! Most likely, you would at first predict that the sole product of the reaction would be the first one shown above, 3-chloro-2,2-dimethylbutane. Why does the second product (2-chloro-2,3-dimethylbutane) form also?
The answer lies in the observation that formation of carbocations is sometimes accompanied by a structural rearrangement. Such rearrangements take place by a shift of a neighboring alkyl group or hydrogen, and are favored when the rearranged carbocation is more stable than the initial carbocation. As you can see in the mechanism below, this holds true in the case of the 3,3-dimethyl-1-butene addition, and explains why the 2-chloro product is actaully the major product: more of it forms than the minor 3-chloro product.
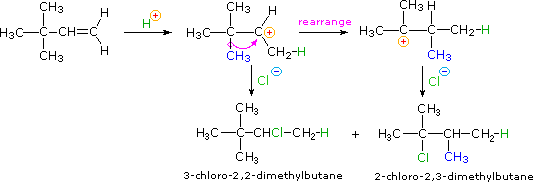
Protonation of the alkene leads to a secondary carbocation, but a methyl shift to a lower-energy tertiary carbocation occurs faster than nucleophilic attack by chlorine, so that when chlorine does attack it does so at carbon #3, rather than carbon #2.
In most examples of carbocation rearrangements that you are likely to encounter, the shifting species is a hydride or methyl group. However, pretty much any alkyl group is capable of shifting. Sometimes, the entire side of a ring will shift over in a ring-expanding rearrangement. Consider the following electrophilic addition of HBr to an alkene: right off the bat, it is very hard to see what is going on here.

Taking into account the possibility of rearrangement, however, we can see how the observed product forms.
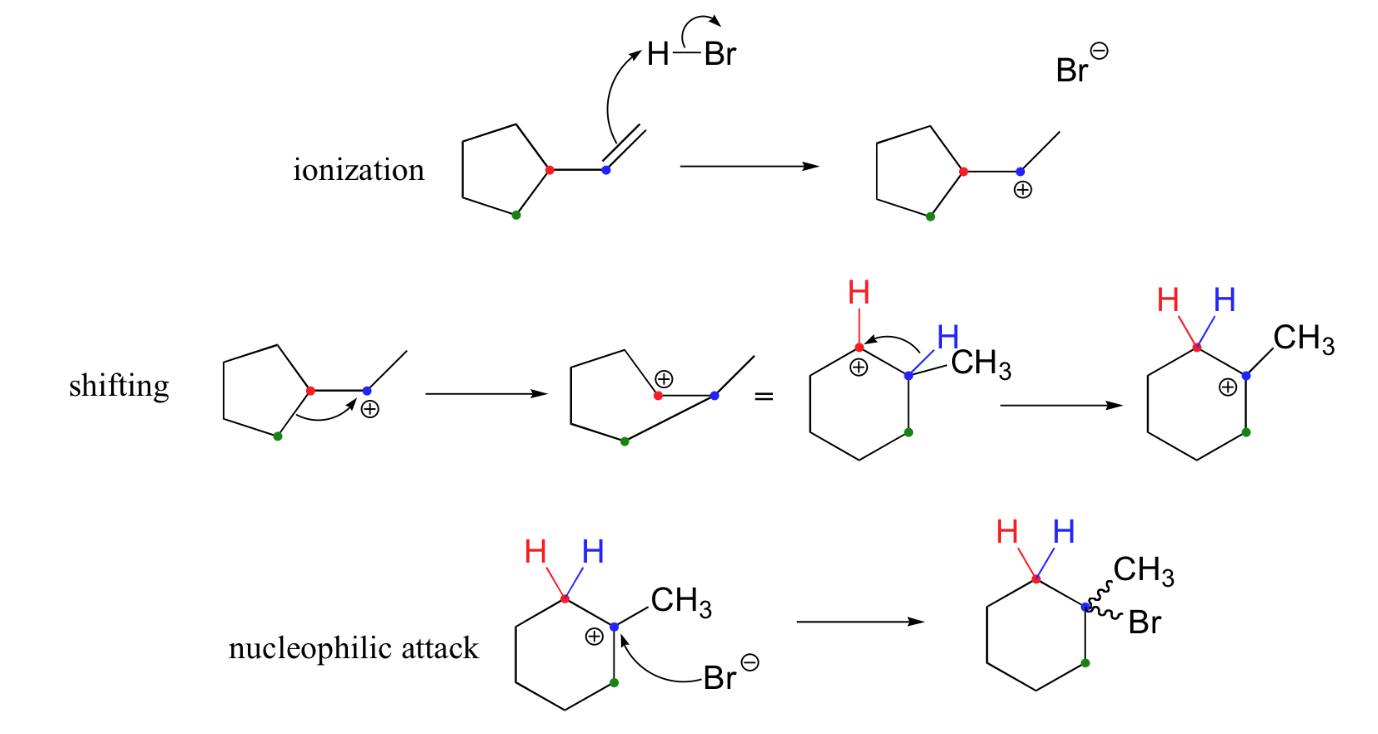
The first 1,2-alkyl shift is driven by the expansion of a five-membered ring to a six-membered ring, which has slightly less ring strain. A hydride shift then converts a secondary carbocation to a tertiary carbocation, which is the electrophile ultimately attacked by the bromide nucleophile.
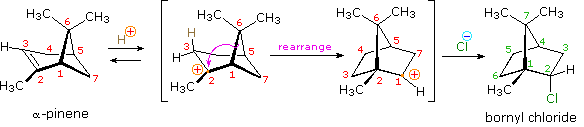
Another factor that may induce rearrangement of carbocation intermediates is strain. The addition of HCl to α-pinene, the major hydrocarbon component of turpentine, gives the rearranged product, bornyl chloride, in high yield. This rearrangement converts a 3º-carbocation to a 2º-carbocation, a transformation that is normally unfavorable. However, the rearrangement also expands a strained four-membered ring to a much less-strained five-membered ring, and this relief of strain provides a driving force for the rearrangement.
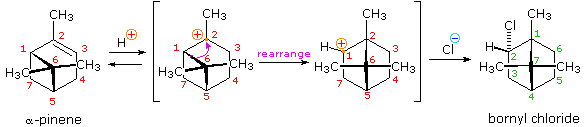
It may be easier to see when shown in a 3-dimensional structure:
- Thuy Hoang
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
Prof. Steven Farmer (Sonoma State University)