
Lecture Extra: Hartree vs. Hartree-Fock, SCF, and Koopman's Theorem
- Page ID
- 40113
Last lecture address two unique aspects of electrons: spin and indistinguishability and how they couple into describing multi-electron wavefunctions. The spin results in an angular momentum that follows the same properties of orbital angular moment including commutators and uncertainty effect. The Slater determinant wavefunction was introduced as a way to consistently address both properties.
The Unsolvable System
The generic multielectron atom including terms for the additional electrons with a general charge \(Z\); e.g.
\[V_{nuclear-electron}(r_1) = -\dfrac {Z}{\left\vert\mathbf{r} - \mathbf{R}\right\vert} \label{8.3.1}\]
in atomic units with \(\left\vert\mathbf{r} - \mathbf{R}\right\vert\) is the distance between the electron and the nucleus, The Hamiltonian must also have terms for electron-electron repulsion (also in atomic units)
\[V_{electron-electron}(r_{12}) = \dfrac {1}{\left\vert \mathbf{r} - \mathbf{r}^{\prime} \right\vert} \label{8.3.2}\]
with \(\left\vert \mathbf{r} - \mathbf{r}^{\prime} \right\vert\) is the distance between electron 1 and electron 2. So the proper multi-electron Hamiltonian can be constructed
\[\hat {H} (r_1, r_2, ... r_n) = -\dfrac {\hbar ^2}{2m_e} \sum _i \nabla ^2_i + \sum _i V_{nuclear-electron} (r_i) + \sum _{i \ne j} V_{electron-electron} (r_{ij}) \label{8.3.3}\]
Given what we have learned from the previous quantum mechanical systems we’ve studied, we predict that exact solutions to the multi-electron Schrödinger equation would consist of a family of multi-electron wavefunctions, each with an associated energy eigenvalue. These wavefunctions and energies would describe the ground and excited states of the multi-electron atom, just as the hydrogen wavefunctions and their associated energies describe the ground and excited states of the hydrogen atom. We would predict quantum numbers to be involved, as well.
The fact that electrons interact through their electron-electron repulsion (final term in Equation \(\ref{8.3.3}\)) means that an exact wavefunction for a multi-electron system would be a single function that depends simultaneously upon the coordinates of all the electrons; i.e., a multi-electron wavefunction:
\[|\Psi (r_1, r_2, \cdots r_i) \rangle \label{8.3.4}\]
Unfortunately, the electron-electron repulsion terms make it impossible to find an exact solution to the Schrödinger equation for many-electron atoms.
The Hartree Approximation
The method for finding best possible one-electron wavefunctions that was published by Hartree in 1948 and improved two years later by Fock. For the Schrödinger equation to be solvable, the variables must be separable. The variables are the coordinates of the electrons. To separate the variables in a way that retains information about electron-electron interactions, the electron-electron term (Equation \(\ref{8.3.1}\)) must be approximated so it depends only on the coordinates of one electron. Such an approximate Hamiltonian can account for the interaction of the electrons in an average way. The exact one-electron eigenfunctions of this approximate Hamiltonian then can be found by solving the Schrödinger equation. These functions are the best possible one-electron functions.
The Hartree approximation starts by invoking an initial ansatz that the multi-electron wavefucntion in Equation \(\ref{8.3.4}\) can be expanded as a product of single-electron wavefunctions
\[\Psi(\mathbf{r}_1,\mathbf{r}_2, \ldots, \mathbf{r}_N) \approx \psi_{1}(\mathbf{r}_1)\psi_{2}(\mathbf{r}_2) \ldots \psi_{N}(\mathbf{r}_N) \label{2.3}\]
from which it follows that the electrons are independent, and interact only via the mean-field Coulomb potential. This yields one-electron Schrödinger equations of the form
\[-\dfrac{\hbar^{2}}{2m} \nabla^{2}\psi_{i}(\mathbf{r}) + V(\mathbf{r})\psi_{i}(\mathbf{r}) = \epsilon_{i}\psi_{i}(\mathbf{r}) \label{2.4}\]
or
\[H_e(r) \psi_{i}(\mathbf{r}) = \epsilon_{i}\psi_{i}(\mathbf{r}) \label{2.4B}\]
where \(V(r)\) is the potential in which the electron moves; this includes both the nuclear-electron interaction
\[V_{nucleus}(\mathbf{r}) = -Ze^{2}\sum_{R} \dfrac{1}{\left\vert\mathbf{r} - \mathbf{R}\right\vert} \label{2.5}\]
and the mean field arising from the \(N-1\) other electrons. We smear the other electrons out into a smooth negative charge density \(\rho(\mathbf{r}')\) leading to a potential of the form
\[V_{electron}(\mathbf{r}) = -e\int d\mathbf{r}^{\prime} \rho(\mathbf{r}^{\prime}) \dfrac{1}{\left\vert \mathbf{r} - \mathbf{r}^{\prime} \right\vert} \label{2.6}\]
where
\[\rho(\mathbf{r}) = \sum_{i}^{\text{occupied}}\vert\psi(\mathbf{r})\vert^{2}.\]
The sum over runs over all occupied states.
The wavefunctions that from this approach with the Hamiltonian \(H_e(r)\) involve possess three kinds of energies:
Kinetic Energy
The Kinetic energy of the electron has an average value is computed by taking the expectation value of the kinetic energy operator \[-\dfrac{\hbar^2}{2m} \nabla^2\] with respect to any particular solution \(\phi_j(r)\) to the Schrödinger equation: \[KE = \langle\phi_j| -\dfrac{\hbar^2}{2m} \nabla^2 |\phi_j\rangle \]
Nuclear-Electron Coulombic Attraction Energy
Coulombic attraction energy with the nucleus of charge \(Z\): \[\langle\phi_j| \dfrac{-Z e^2}{\left\vert\mathbf{r} - \mathbf{R}\right\vert} |\phi_j\rangle\]
Electron-Electron Coulombic Repulsion Energy
Coulomb repulsion energies with all of the \(N-1\) other electrons, which are assumed to occupy other atomic orbitals denoted \(\phi_K\), with this energy computed as
\[\sum_{J\neq K} \langle\phi_j(r) \phi_K(r’) |\dfrac{e^2}{|r-r’|} | \phi_j(r) \phi_K(r’)\rangle.\label{8.3.8}\]
The Dirac notation \(\langle\phi_j(r) \phi_K(r’) |\dfrac{e^2}{|r-r’|} | \phi_j(r) \phi_K(r’)\rangle\) is used to represent the six-dimensional Coulomb integral
\[J_{J,K} = \int |\phi_j(r)|^2 |\phi_K(r’)|^2 \dfrac{e^2}{r-r'} dr dr’ \label{8.3.9}\]
that describes the Coulomb repulsion between the charge density \(|\phi_j(r)|^2\) for the electron in \(\phi_j\) and the charge density \(|\phi_K(r’)|^2\) for the electron in \(\phi_K\). Of course, the sum over \(K\) must be limited to exclude \(K=J\) to avoid counting a “self-interaction” of the electron in orbital \(\phi_j\) with itself.
The total energy \(\epsilon_J\) of the orbital \(\phi_j\), is the sum of the above three contributions:
\[\epsilon_J = \langle\phi_j| - \dfrac{\hbar^2}{2m} \nabla^2 |\phi_j\rangle + \langle\phi_j| \dfrac{-Z e^2}{\left\vert\mathbf{r} - \mathbf{R}\right\vert} |\phi_j\rangle + \sum_{J\neq K} \langle\phi_j(r) \phi_K(r’) |\dfrac{e^2}{|r-r’|} | \phi_j(r) \phi_K(r’)\rangle.\label{8.3.10}\]
This treatment of the electrons and their orbitals is referred to as the Hartree-level of theory.
When screened hydrogenic atomic orbitals are used to approximate the \(\phi_j\) and \(\phi_K\) orbitals, the resultant \(\epsilon_J\) values do not produce accurate predictions. For example, the negative of \(\epsilon_J\) should approximate the ionization energy for removal of an electron from the orbitals \(\phi_j\). Such ionization potentials (IP s) can be measured, and the measured values do not agree well with the theoretical values when a crude screening approximation is made for the atomic orbitals.
The Self-Consistant Field (SCF) Approach to the Variational Method
An important unsolved problem in quantum mechanics is how to deal with indistinguishable, interacting particles - in particular electrons which determine the behavior of almost every object in nature. The basic problem is that if particles interact, that interaction must be in the Hamiltonian. So until we know where the particles are, we cannot write down the Hamiltonian, but until we know the Hamiltonian, we can’t tell where the particles are.
The idea is to solve the Schrödinger equation for an electron moving in the potential of the nucleus and all the other electrons. We start with a guess for the trial electron charge density, solve N/2 one-particle Schrödinger equations (initially identical) to obtain N electron wavefunctions. Then we construct the potential for each wavefunction from that of the nucleus and that of all the other electrons, symmetrize it, and solve the N/2 Schrödinger equations again. This method is ideal for a computer, because it is easily written as an algorithm Figure \(\PageIndex{1}\).
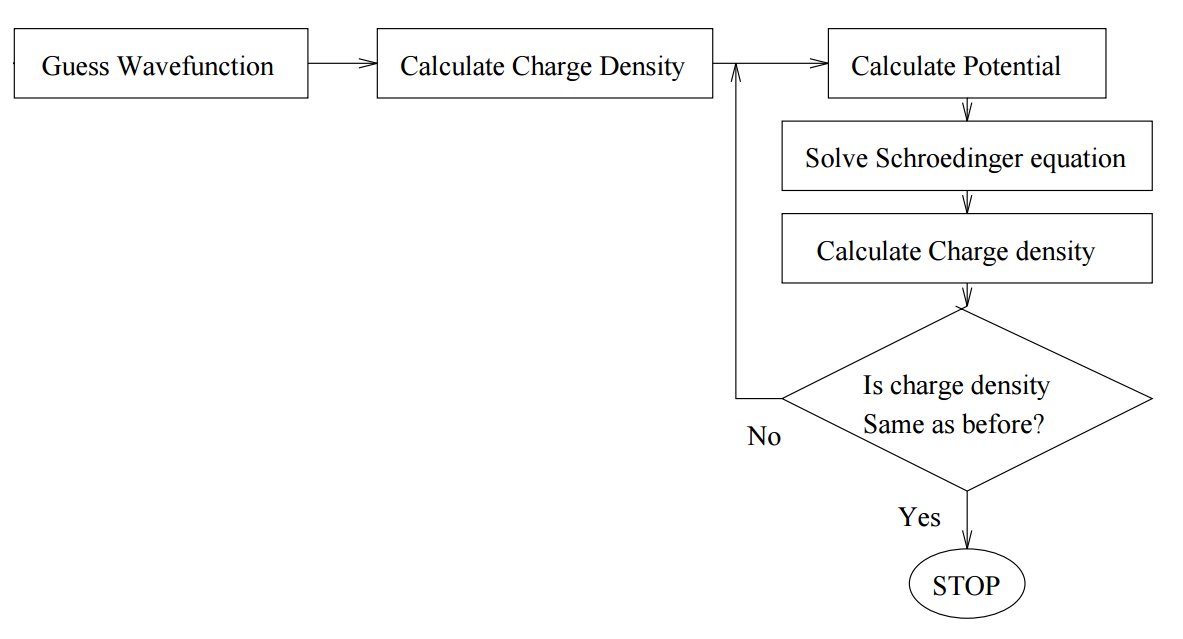
Although we are concerned here with atoms, the same methodology is used for molecules or even solids (with appropriate potential symmetries and boundary conditions). This is a variational method, so wherever we refer to wavefunctions, we assume that they are expanded in some appropriate basis set.
Hartree-Fock method
Fock improved on Hartree’s method by using proper "antisymmetrized wavefunctions" (called the Hartree-Fock method) instead of simple one-electron wavefunctions. The generalized Slater determinant for a multi-electrom atom with \(N\) electrons is then
\[ \psi(\mathbf{r}_1, \mathbf{r}_2, \ldots, \mathbf{r}_N)=\dfrac{1}{\sqrt{N!}} \left| \begin{matrix} \chi_1(\mathbf{r}_1) & \chi_2(\mathbf{r}_1) & \cdots & \chi_N(\mathbf{r}_1) \\ \chi_1(\mathbf{r}_2) & \chi_2(\mathbf{r}_2) & \cdots & \chi_N(\mathbf{r}_2) \\ \vdots & \vdots & \ddots & \vdots \\ \chi_N(\mathbf{r}_N) & \chi_2(\mathbf{r}_N) & \cdots & \chi_N(\mathbf{r}_N) \end{matrix} \right| \label{5.6.96}\]
If we go through the same process as above with this more proper wavefunction we get similar, but not identical single-particle Hartree-Fock equations:
\[-\dfrac{\hbar^{2}}{2m}\nabla^{2}\psi_{i}(\mathbf{r}) + V_{nucleus}(\mathbf{r})\psi_{i}(\mathbf{r}) + V_{electron}(\mathbf{r})\psi_{i}(\mathbf{r}) - \sum_{j} \int d\mathbf{r}^{\prime} \dfrac{\psi^{\star}_{j}(\mathbf{r}') \psi^{\star}_{i}(\mathbf{r}') \psi_{j}(\mathbf{r}) } {\left \vert \mathbf{r} - \mathbf{r}^{\prime} \right\vert} = \epsilon_{i}\psi_{i}(\mathbf{r}). \label{2.9}\]
can be recast as series of Schrödinger-like equations:
\[ \hat {F} | \varphi _i \rangle = \epsilon _i| \varphi _i \rangle \label {8.7.2}\]
where
- \(\hat {F}\) is called the Fock operator and
- \( \{| \varphi_i \rangle \}\) are the Hatree-Fock orbitals with corresponding energies \(\epsilon_i\).
The Fock operator is a one-electron operator and solving a Hartree-Fock equation gives the energy and Hartree-Fock orbital for one electron.
The nature of the Fock operator reveals how the Hartree-Fock (HF) or Self-Consistent Field (SCF) Method accounts for the electron-electron interaction in atoms and molecules while preserving the idea of independent atomic orbitals. The wavefunction written as a Slater determinant of spin-orbitals is necessary to derive the form of the Fock operator, which is
\[\hat {F} = \hat {H} ^0 + \sum _{j=1}^N ( 2 \hat {J} _j - \hat {K} _j ) = -\dfrac {\hbar ^2}{2m} \nabla ^2 - \dfrac {Ze^2}{4 \pi \epsilon _0 r} + \sum _{j=1}^N (2\hat {J}_j - \hat {K} _j ) \label {8.7.3}\]
- \(\hat{J}\) is the Coulomb operator, defining the electron-electron repulsion energy due to each of the two electrons in the jth orbital.
- \(\hat {K}\) is the exchange operator, defining the electron exchange energy due to the antisymmetry of the total n-electron wave function. This (so called) "exchange energy" operator, K, is simply an artifact of the Slater determinant.
The Hartree-Fock equations \(h_e \phi_i = \epsilon_i \phi_i\) imply that the orbital energies \(\epsilon_i\) can be written as:
\[\epsilon_i = \langle \phi_i | h_e | \phi_i \rangle = \langle \phi_i | T + V | \phi_i \rangle + \sum_{j({\rm occupied})} \langle \phi_i | J_j - K_j | \phi_i \rangle \label{8.7.6}\]
\[= \langle \phi_i | T + V | \phi_i \rangle + \sum_{j({\rm occupied})} [ J_{i,j} - K_{i,j} ],\label{8.7.7}\]
where \(T + V\) represents the kinetic (\(T\)) and nuclear attraction (\(V\)) energies, respectively. Thus, \(\epsilon_i\) is the average value of the kinetic energy plus Coulombic attraction to the nuclei for an electron in \(\phi_i\) plus the sum over all of the spin-orbitals occupied in \(\psi\) of Coulomb minus Exchange interactions of these electrons with the electron in \(\phi_i\).
Hartree vs. Hartree-Fock
The many-body wave function gives the N-particle distribution function, i.e. \(|Φ(r_1, ..., r_N )|^2\) is the probability density that particle 1 is at \(r_1\), ..., and particle \(N\) is at \(r_N\).
However, when trying to work out the interaction between electrons, what we want to know is the probability of finding an electron at \(r\), given the positions of all the other electrons \(\{r_i\}\). This implies that the electron behaves quantum mechanically when we evaluate its wavefunction, but as a classical point particle when it contributes to the potential seen by the other electrons.
Hartree-Fock is not that bad... but not super great either
| Atom | Hartree-Fock Energy | Experiment |
|---|---|---|
| \(He\) | \(-5.72\) | \(-5.80\) |
| \(Li\) | \(-14.86\) | \(-14.96\) |
| \(Ne\) | \(-257.10\) | \(-257.88\) |
| \(Ar\) | \(-1053.64\) | \(-1055.20\) |
Koopman's Theorem
Koopmans' theorem states that the first ionization energy is equal to the negative of the orbital energy of the highest occupied molecular orbital. Hence, the ionization energy required to generated a cation and detached electron is represented by the removal of an electron from an orbital without changing the wavefunctions of the other electrons. this is called the "frozen orbital approximation."
Within this model, the energy difference between the daughter and the parent can be written as follows (\(\phi_k\) represents the \(k^{th}\) spin-orbital that is added or removed):
Hence for electron detachment (ionization):
\[E_{N-1} - E_N = - \epsilon_k \]
For electron attachement (electron affinity): It is sometimes claimed that Koopmans' theorem also allows the calculation of electron affinities as the energy of the lowest unoccupied molecular orbitals (LUMO) of the respective systems. However, Koopmans' original paper makes no claim with regard to the significance of eigenvalues other than that corresponding to the HOMO. Nevertheless, it is straightforward to generalize the original statement of Koopmans' to calculate the electron affinity in this sense.
\[E_N - E_{N+1} = - \epsilon_{k+1} .\]
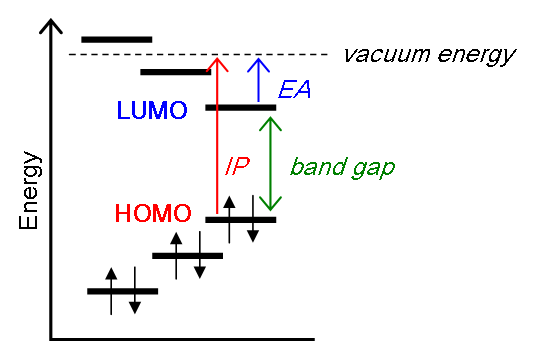
Koopman's Theorem in action
Koopman's Theorem argues that you do not need to perform two independent quantum calculations to determine the ionization energy and electron affinity as done in the ab initio homeworks, but only one is needed and the energies of the spin-orbitals are needed.
Contributors and Attributions
- Wikipedia
David M. Hanson, Erica Harvey, Robert Sweeney, Theresa Julia Zielinski ("Quantum States of Atoms and Molecules")
facultystaff.richmond.edu/~s...Ch_13c_S11.pdf
www.thphys.may.ie/Notes/MP463/MP463_Ch4.pdf
www.clt.astate.edu/sreeve/Mul...on%20Atoms.pdf