
The Edman Degradation
- Page ID
- 165171
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)After completing this section, you should be able to
- describe how an Edman degradation is used to determine the sequence of the amino acid residues in peptides containing up to 20 such residues.
- describe, briefly, how the procedure is modified to deal with peptides and proteins containing more than 20 amino acid residues.
- write a detailed mechanism for the Edman degradation.
- determine the structure of a peptide, given a list of the fragments that are produced by a partial acid hydrolysis.
- determine the structure of a peptide, given a list of the fragments that are produced when the peptide is cleaved by a specific enzyme and the details of the types of bonds cleaved by that enzyme.
- predict the fragments that would be produced when a peptide of known structure is cleaved by a specific enzyme, given sufficient information about the types of bonds that are cleaved by the enzyme in question.
Make certain that you can define, and use in context, the key term below.
- Edman degradation
The reagent used in the Edman degradation is phenyl isothiocyanate. You may find it helpful to review the relationship between cyanates, isocyanates, thiocyanates and isothiocyanates.

You need not memorize the specific peptide bonds that are broken by the enzymes trypsin and chymotrypsin.
Edman degradation is the process of purifying protein by sequentially removing one residue at a time from the amino end of a peptide. To solve the problem of damaging the protein by hydrolyzing conditions, Pehr Edman created a new way of labeling and cleaving the peptide. Edman thought of a way of removing only one residue at a time, which did not damage the overall sequencing. This was done by adding Phenyl isothiocyanate, which creates a phenylthiocarbamoyl derivative with the N-terminal. The N-terminal is then cleaved under less harsh acidic conditions, creating a cyclic compound of phenylthiohydantoin PTH-amino acid. This does not damage the protein and leaves two constituents of the peptide. This method can be repeated for the rest of the residues, separating one residue at a time.
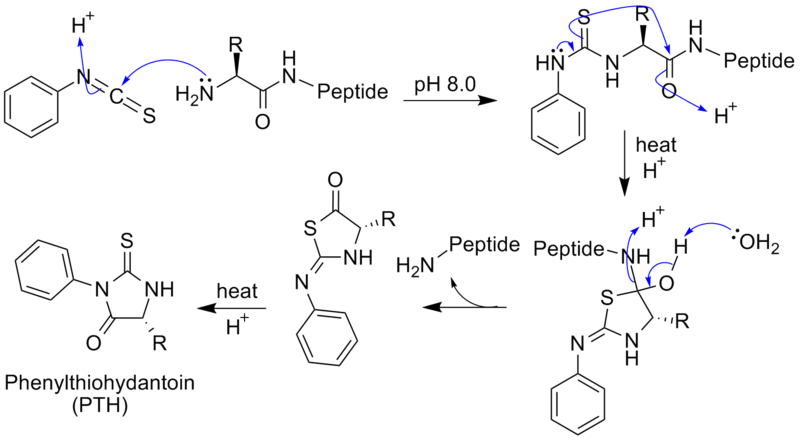
Edman degradation is very useful because it does not damage the protein. This allows sequencing of the protein to be done in less time. Edman sequencing is done best if the composition of the amino acid is known. As we saw in Section 26.5, to determine the composition of the amino acid, the peptide must be hydrolyzed. This can be done by denaturing the protein and heating it and adding HCl for a long time. This causes the individual amino acids to be separated, and they can be separated by ion exchange chromatography. They are then dyed with ninhydrin and the amount of amino acid can be determined by the amount of optical absorbance. This way, the composition but not the sequence can be determined
Sequencing Larger Proteins
Larger proteins cannot be sequenced by the Edman sequencing because of the less than perfect efficiency of the method. A strategy called divide and conquer successfully cleaves the larger protein into smaller, practical amino acids. This is done by using a certain chemical or enzyme which can cleave the protein at specific amino acid residues. The separated peptides can be isolated by chromatography. Then they can be sequenced using the Edman method, because of their smaller size.
In order to put together all the sequences of the different peptides, a method of overlapping peptides is used. The strategy of divide and conquer followed by Edman sequencing is used again a second time, but using a different enzyme or chemical to cleave it into different residues. This allows two different sets of amino acid sequences of the same protein, but at different points. By comparing these two sequences and examining for any overlap between the two, the sequence can be known for the original protein.
For example, trypsin can be used on the initial peptide to cleave it at the carboxyl side of arginine and lysine residues. Using trypsin to cleave the protein and sequencing them individually with Edman degradation will yield many different individual results. Although the sequence of each individual cleaved amino acid segment is known, the order is scrambled. Chymotrypsin, which cleaves on the carboxyl side of aromatic and other bulky nonpolar residues, can be used. The sequence of these segments overlap with those of the trypsin. They can be overlapped to find the original sequence of the initial protein. However, this method is limited in analyzing larger sized proteins (more than 100 amino acids) because of secondary hydrogen bond interference. Other weak intermolecular bonding such as hydrophobic interactions cannot be properly predicted. Only the linear sequence of a protein can be properly predicted assuming the sequence is small enough.
Contributors and Attributions
- Wikibooks (Structural Biopchemistry). The content on this page is licensed under a CC-SA-BY 3.0 license.