
2.1: Chemical Representations on Computer Part I
- Page ID
- 83686

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)
 |
UALR 4399/5399: ChemInformatics |
 |
Learning Objectives:
- Describe and be able to identify ambiguous, unambiguous, and canonical representations of chemical structure, as well as explicit and implicit information contained in these representations.
- Describe each of the four major approaches to machine representation of chemical structure (connection tables, graphic visualizations, line notation, and descriptive representations), as well as the advantages and drawbacks of each of these forms.
- Describe how database record IDs relate to representations of chemical structure.
- Describe lookup and translation approaches to exchanging chemical identifiers, including what countertranslation is and why it can be useful.
CHEMICAL REPRESENTATION FOR CHEMINFORMATICS
Most often, data and information about chemical compounds is either directly about molecular structure (for example, a 2D structural formula, or 3D atomic coordinates for a particular conformation of a compound), or is tied to a molecular structure (for example, physical properties of a compound, which you identify by its structural formula). The notion of indexing, sorting, searching and retrieving information using molecular structures originated within the domain of modern chemistry.
Almost all chemists engage in communication tasks to register, search, view, and publish molecular structures. Most forms of chemical representation were developed with these uses in mind. Cheminformatics involves storing, finding, and analyzing these structures using the data-processing power of computers to match chemical compounds with literature publications, measured properties, synthetic procedures, spectra, and computational studies. To do this work, computers need to use chemical representation to identify, exchange and validate information about chemical compounds.
In order for (human) chemists to rely on insights from cheminformatics, it is important to understand the way in which computers store and analyze chemical structure, the methods that computer programs employ, and the results that they produce. Therefore, cheminformatics depends upon the use of representations of molecular structures and related data that are understandable both to human scientists and to machine algorithms.
FORMULATING CHEMICAL STRUCTURE DATA
Interacting with a machine is a form of communication. How does communication between chemists differ from communication between a chemist and a machine? In cheminformatics, you are working within a system governed by strict rules that are explicitly defined. If you know the rules, then you can make the system work for you. If you don't know the rules for a given form of representation, sometimes features designed to satisfy the requirements of one context will appear as bugs in another context.
If one chemist was to recommend to another that a reaction should be performed using "chloroform" as a solvent for a reaction, this would generally be a successful exercise in communication. For all practical purposes, this word is understood by every chemist, and has no ambiguity. However, because "chloroform" is a so-called trivial name, there is no formula for converting it into the actual chemical structure that it represents, and a machine will not be able to participate in this exchange of information unless it has been explicitly instructed as to the chemical structure that this word represents, expressed in a format that the machine can work with.
A more descriptive way to communicate the composition that is chloroform is by chemical formula, in this case CHCl3. A computer program could interpret basic molecular structure rules to determine that the substance being described has 5 atoms: 1 carbon, 1 hydrogen and 3 chlorine. Assembling this into a molecule with bonds can be based on valence rules, identifying 4 of the atoms as normally monovalent and one as normally tetravalent. It is quite simple to create a software algorithm that can join the atoms together in the most obvious way, which also happens to be correct.
Beyond such tiny simple molecules, difficulties soon arise. Some of these ambiguities affect human chemists in the same way that they affect machines. Consider the molecular formula of C3H6O, which is associated with multiple reasonable structures, including a ketone, an aldehyde, a cyclic alcohol, oxygenated alkenes and cyclic ethers, one of which exists as two enantiomers:
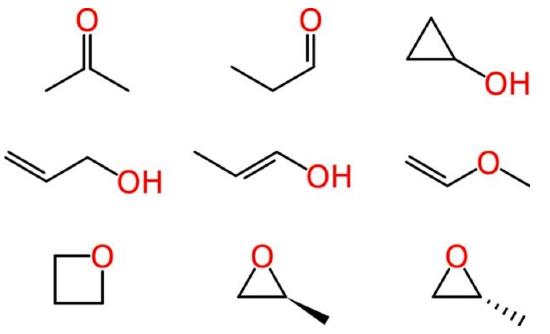
Ambiguous representations can refer to more than one chemical entity. This is true of most chemical names when used unsystematically, such as “octane,” when employed as a common term for all saturated hydrocarbons with eight carbon atoms, rather than systematically to indicate the straight-chain isomer only. Empirical and molecular formulas are also typically ambiguous.
In an unambiguous system of representation, each name or formula refers to exactly one chemical entity, typically in a way that allows you to draw a structural formula for it. However, each chemical entity might be represented by more than one name or formula. A canonical form is a completely unique representation within a system. For example, “diethyl ketone” and “3-pentanone” are both unambiguous names: each represents one and only one compound. However, since they represent the same compound, they are not unique names. Within the system of Preferred IUPAC Names (see below), “3-pentanone” is a canonical name – an unambiguous and unique representation of this compound.
Note that, since canonical names are necessarily canonical within a system, they might not function properly if you are interested in structural information that is not addressed within the system, or if you do not have structural information that is required by the system. For example, within a system that does not address stereochemistry, the different enantiomers of a chiral compound will have the same “canonical” representation. Within a system that requires the specification of stereochemistry, on the other hand, you will have to choose between stereospecific canonical representations. If you happen to be working with a racemic mixture or a compound of unknown stereo configuration, this may lead to misrepresentation and misunderstanding.
A chemical structure representation contains two kinds of information: explicit and implicit. [H1] Explicit information is what’s directly represented in a data structure and should at minimum contain what otherwise would not be known, such as the specific atom in a carbon skeleton to which a substituent is attached. Implicit information is what you (or a computer) can figure out from a data structure, given some knowledge of general principles and a little bit of work.
In general, data structures that contain less explicit information are more simple and compact, but they require more computation to draw chemical conclusions from them. Data structures that contain more explicit information take up more space and are at greater risk of containing inconsistencies, but they can be more quickly analyzed in a wider variety of ways.
To automate functions on chemical data, the data structure needs to be systematically defined and consistently applied. These definitions are part of what constitutes explicit information that an algorithm can readily identify and parse. Balancing the level of explicit information can also impact the ambiguity of a system, and the ability to accurately exchange chemical structures between systems. These are especially important considerations for operations that range across a significant portion of the corpus of reported chemical compounds (well over 100 million), beyond the scale at which human validation of results is possible.
REPRESENTATING CHEMICAL STRUCTURE DATA
Generally, the most effective way to communicate with another chemist about the structure of a compound is to draw its structural formula. A structural formula is any formula that indicates the connectivity of a compound – that is, which of its atoms are linked to each other by covalent bonds.
It just so happens that structural formulas can be fairly directly mapped to a computer-friendly data structure: a molecular graph stored as a connection table. Connection tables do for computers what systematic nomenclature does for human chemists: they the organize structural information defined in a molecular graph in a form that is easier to read and to order in a list. The difference is that computers can read, sort, search, and group connection tables far faster than humans can work with systematic names or any other kind of formula or notation. Connection tables are covered in more depth in the second part of this module.
Chemical structure is represented on computers in several forms, usually generated from chemical connectivity data stored in connection tables. These representations are designed to facilitate many human and computer functions and are machine-actionable as long as they can be tied into a database of connection tables or an algorithm for translating a given representation into a connection table. Besides connection tables, the most common forms of machine-readable representations are graphic visualizations, line notations, and other descriptive forms such as nomenclature.
Graphic Visualizations
Chemists most frequently think about chemical structure in 2D, and molecules actually exist in 3D physical space. Most chemical data systems offer 2D and 3D visualizations that human chemists can use to communicate preferences for searching and analysis. The 2D coordinates stored in a connection table can be used to infer and display chemical information, including the basic structural formula and additional information such as the E/Z geometry of alkene-like double bonds and the cis/trans isomerism of ligands in a square planar metal complex or substituents on a cyclic alkane. 2D representations are designed to mimic the experience of drawing structural formulas on paper. Human users can further fix these electronic drawings as images to use in publications and presentations, but these image files are no longer connected directly to chemical data and are thus not machine readable.
3D (x,y,z) coordinates can also be stored for each atom and used to display the conformation of a molecule. These coordinates may be determined experimentally (typically via x-ray crystallography), or calculated (using force-fields, quantum chemistry, molecular dynamics or composite models such as docking). Understanding a molecule's actual shape, whether it be in solution, in a vacuum, or in the binding site of a protein, opens up a whole new domain of computational chemistry. Most molecules have some flexibility, and even if a given conformation is the most stable, there are often a number of competing shapes to consider. Knowing how a particular set of coordinates was determined is crucial to making intelligent use of it for cheminformatics purposes.
Machine generation and interpretation of graphical representations involve persistent challenges that can affect the accuracy of chemical information communicated between humans and machines. Because structural formulas were originally invented to represent organic compounds, both people and computer programs will tend to assume, as a default, that structural formulas represent networks of atoms linked by discrete covalent bonds. This chemical logic makes it possible for machines to handle these graphical representations, and many chemistry databases are organized around this principle of explicit connectivity. However, delocalized systems, non-covalent molecules such as coordination compounds, and other classes of chemical substances do not fit easily into these conventions for generating and interpreting graphical representations. There are different practices among chemists for depicting such structural features, and there are no broadly accepted conventions for representing them in connection tables.
For example, the IUPAC standards for graphical representation in publications specify the use of curved circle bonds instead of alternating single and double bonds for the pi-system within aromatic rings. They also indicate that coordination bonds between a metal and an aromatic system should be represented as a single bond from the metal into the middle of the ring, and that formal charges should not be shown. However, these conventions are beyond the scope of basic rules of valence and are difficult to program consistently. As a result, coordination compounds represented in this way may be captured as separate fragments in connection tables. Computer software may interpret the end of the bond in the middle of an aromatic ring as an implied additional methyl group. Circles within rings may not be decipherable in a computer program, and the associated electron system may be ignored entirely. A more common representation of coordination compounds used in chemistry databases addresses these problems by including explicit bonds between the metal and each atom in the ring. This follows basic rules of valence and enables a more consistent approach to structure search. However, this notation can be misleading for human readers as the nature of the association between the metal and the ring is not covalent bonding.
Line Notations
Line notations represent chemical structures as a linear string of symbolic characters that can be interpreted by systematic rule sets. They are widely used in Cheminformatics because a) many computational processes operate more effectively on data structured as linear strings than data structured as tables, and b) line notations can be reasonably legible to human chemists designing functions with these tools. Linear representations are particularly well-suited to many identification and characterization functions, such as determining:
- whether molecules are the same;
- how similar they are, according to some metric;
- whether one molecular entity is a substructure of another;
- whether two molecules are related by a specific transformation;
- what happens when molecules are cut into pieces and grafted together at different positions.
In these and other applications of cheminformatics, linear representations have key advantages for speed and automation, especially when you’d like to handle huge numbers of structures (e.g. searching a large database).
Examples of line notations include the Wiswesser Line-Formula Notation (WLN), Sybyl Line Notation (SLN) and Representation of structure diagram arranged linearly (ROSDAL). Currently, the most widely used linear notations are the Simplified Molecular-Input Line-Entry System (SMILES) and the IUPAC Chemical Identifier (InChI), which are described in the third part of this module.
Descriptive Representation
Systematic names describe the structural formula of compounds. If you know the rules and vocabulary, you should be able to write a name based on a structural formula and vice-versa. Chemists have developed various ways of translating formulas into names, so it is nearly always possible to write more than one systematic name for a given compound.
IUPAC (International Union of Pure and Applied Chemistry) nomenclature is a well-known international system of chemical names that is generally systematic but flexible, allowing the use of certain well-established trivial names. Since systematic IUPAC names are made according to formalized rules, they could, in principle, be used by both humans and computers. However, IUPAC names are often quite difficult for chemists to read, let alone to write, and the rules are non-canonical, resulting in numerous different options for naming each compound. IUPAC has introduced even more rules for determining canonical Preferred IUPAC Names (PINs) that are oriented toward making systematic names more easily readable by machines.
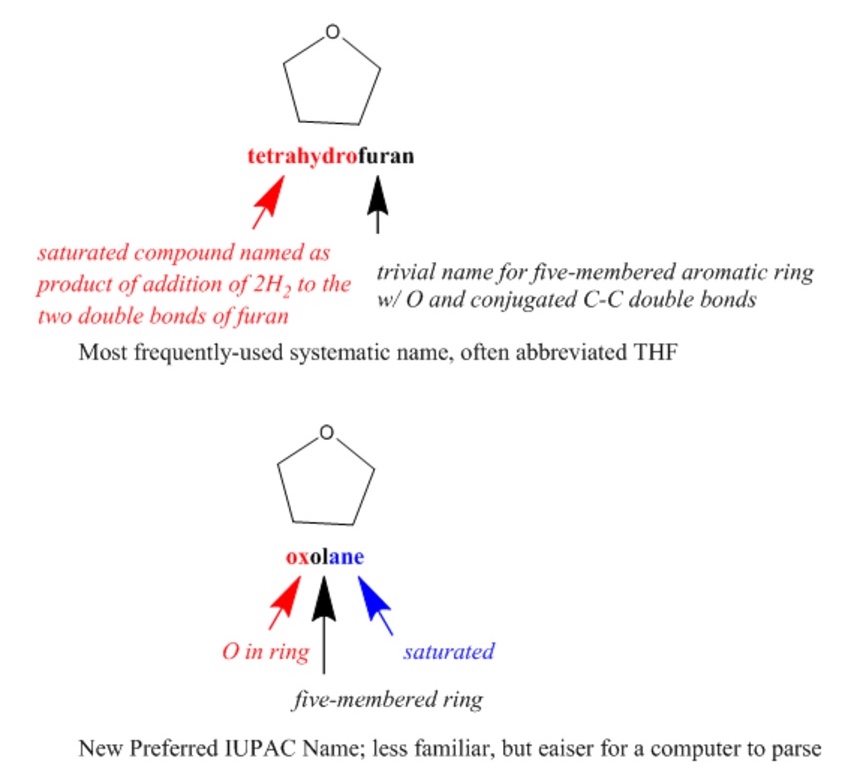
Semantic technologies further enable systematic classification and organization of scientific terms, including descriptions of chemical structures, such as provided by ChEBI (Chemical Entities of Biological Interest). ChEBI describes small molecular entities based on nomenclature, symbolism and terminology endorsed by IUPAC and the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). This dataset is highly curated by both human experts and machine processes, is openly searchable and programmatically accessible, and includes full references to original authoritative sources.
IDENTIFYING CHEMICAL STRUCTURE DATA
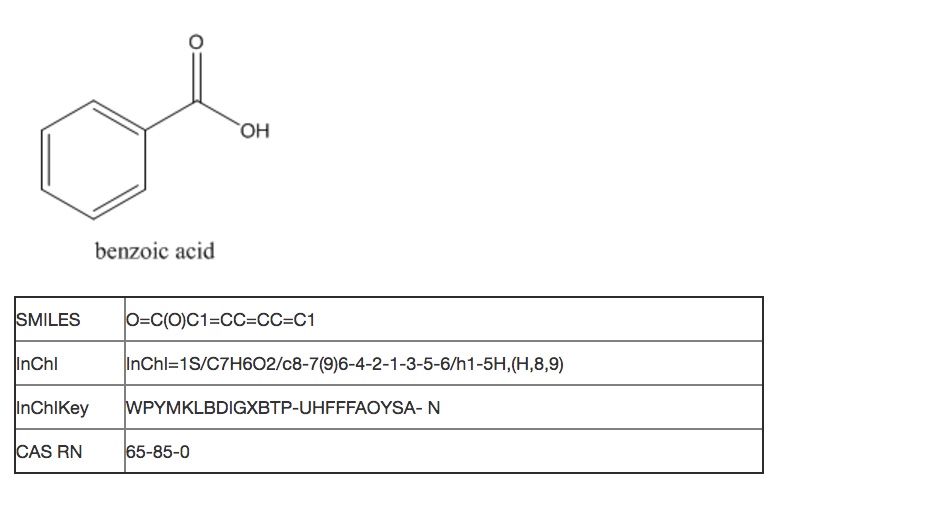
In Cheminformatics, working programmatically and at scale, you need to be able to automatically retrieve, organize and differentiate large numbers of chemical substances by structural data. Specific databases that collect and organize information by chemical substances will usually have a record ID system that identifies the profile of the compound or substance as assembled in that database. However, most record ID systems use alpha-numeric strings that are not defined by chemical structure rules, in contrast to linear structure notations such as SMILES and InChI. Record IDs should not be used as proxies for molecular structure for cheminformatics purposes, unless there is an automated way to look up and retrieve the original structure data files.
The most familiar system of chemical record IDs is the Chemical Abstracts Service Registry Number (CAS RN). The CAS Registry can be searched using CAS products such as SciFinder and STN. In this database, unique identifiers are assigned to each chemical substance reported in the literature, providing an unambiguous way to identify a chemical substance or system within the Registry when there are many possible systematic, generic, proprietary or trivial names. CAS RNs are numeric identifiers that can contain up to ten digits, divided by hyphens into three parts: the first consisting of two to seven digits, the second consisting of two digits, and the third consisting of a single digit. These numbers themselves have no inherent chemical meaning about structure, but are assigned in sequential order to new substances in a variety of forms as they are reported in the literature.
Record IDs can sometimes be considered de facto identifiers for chemicals by the users of these databases. However, these ID systems are specific to their originating data structure and are not necessarily suitable to use in practice to systematically identify compounds outside of these databases. CAS Registry records reflect what has been reported about substances and not necessarily a systematic gestalt of chemical structures. A CAS RN may refer to a substance profile with several structures for a multi-component system, or no structure at all, or an unspecified configuration. While CAS RNs have been widely used, the CAS database is a proprietary and controlled registry. Most CAS RNs that appear online are not verified relative to characterizing information about a substance. It may not be possible to disambiguate whether a CAS RN refers to a single chemical compound or to a component in a mixture, for example.
The PubChem CID and the ChemSpider ID are two other alphanumeric record ID systems that do not inherently contain chemical structure information in the ID notation itself. However, these IDs refer to chemical structural data that is systematically generated and organized within these databases, which can be openly and programmatically searched. Thus PubChem and ChemSpider record IDs are often used by computer programs as links to identify chemical structure data within these large systems. PubChem cheminformatics functionality will be discussed as an example more extensively later in this course.
Even if a record ID is canonical, that does not mean that it identifies a compound or substance with absolute precision. For example, there is a separate CAS RN for each enantiomer of lactic acid, as well as a third RN that might identify either the racemic mixture or the compound with unspecified stereochemistry. How do you know which isomer you have if just a number is given in a report? There is no way to tell algorithmically, and you may need to search with all three RNs and potentially retrieve many false hits if you are interested in only one particular isomer. Similarly, the canonical form of SMILES does not take R/S stereoisomerism into account, so each enantiomer of a compound will have the same canonical SMILES formula.
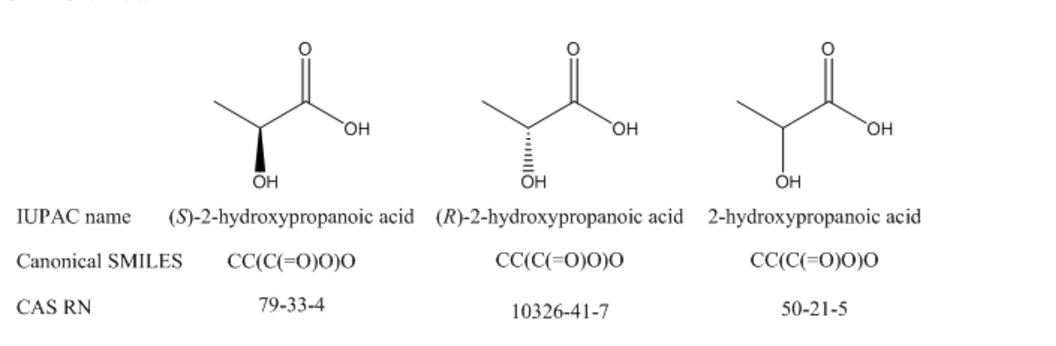
The International Chemical Identifier (InChI) also provides a rule set that generates canonical structure representations, but can be similarly hampered by ambiguity of higher level structure considerations. PubChem and ChemSpider incorporate the InChI algorithm as part of their data validation schemata, and many other databases accept InChI and SMILES strings as queries to search for chemical structures.
InChI can be hashed into a shorter form of 27 characters, called an InChIKey. This allows for even easier searching of general systems such as Google, which can locate chemical structures in many open databases. Once hashed, InChIKeys are not reversible and cannot algorithmically generate a chemical structure, except by looking up an InChIKey in a database record that also contains the structure. Thus the InChIKey can serve to confidentially notate proprietary structural information that has not yet been disclosed.
EXCHANGING CHEMICAL REPRESENTATIONS
Effective aggregation and re-use of chemical data often involves swapping the identifier that you’ve located for another representation for the same compound that’s more convenient for your purpose. For example, if you are interested in comparing the structures of a list of compounds for which you have registry numbers, you need to swap those registry numbers for structural formulas, connection tables, or another sort of representation that gives you the structural information you’re looking for.
This sort of re-use of notation happens a lot in cheminformatics – after all, some kinds of cheminformatics analysis weren’t even conceivable when most common forms of chemical names and formulas first caught on. But the repurposing of notation isn’t unique to cheminformatics. In fact, as long as chemical names and formulas as we know them have been around, chemists have been re-using names, deciding that they fit other purposes better than the ones for which they were intended, or trying to change them in ways that undermine their original purpose.
There are two basic approaches to exchanging chemical identifiers: lookup and translation. In the case of lookup, you locate the identifier that you have in an existing database that lists various different identifiers for each compound, and you select the other identifier that you want. This is like using a thesaurus. There are several tools available for cross-referencing identifiers from different databases, including the CACTUS, UniChem, and PubChem Identifier Exchange services. All of these lookup services accept most linear identifiers as queries.
In the case of translation, you use a set of rules (or a computer uses an algorithm) to take apart one type of representation of a compound and convert it into another type of representation for the same compound. There are several open toolkits available for translation, including RDKit, OpenBabel, Chemistry Development Kit, among others (see Blue Obelisk).
Like words for the same object in different languages, even when two representations are meant to refer to exactly the same compound, they differ in their connotations. They describe different aspects of structure more or less explicitly, they emphasize different kinds of family relationships or functional patterns, and they draw upon different ways of interpreting chemical objects and phenomena. Identifiers are not equally specific: for example, you can translate a structural formula into a single molecular formula, but you cannot translate that molecular formula back into a structural formula.
Translation can thus be quite lossy and lookup may identify a close but not precise enough match for your need. It can be difficult to catch these problems during the exchange and different tools may present different problems. Naoki Sakai, a scholar of translation in literature and politics, has written, “Every translation calls for a countertranslation.” The same is true in chemistry. Can you use a newly generated representation and get back to the original one? When exchanging representation formats, countertranslation can identify what might have gotten lost or inadvertently added in translation.
Large chemical databases use validation and counter-translation as part of standardizing the data included in their chemical records. For example, they may collect data that includes both systematic names and molecular structures and run each of these name-to-structure and structure-to-name conversions to match any previous instances of these compounds in their databases or identify any potential errors.
As you process chemical structure data, consider how usable your output is for a diversity of unknown future cheminformatics applications. Follow common practices such as those used in the large public chemical databases, and carefully document your notation mapping and rules. Whenever you exchange chemical structure data, keep a provenance trail to the original data source and note the tools and resources you have used with the data so that others following on your work can use your data efficiently (including yourself!).
Different forms of chemical notation are more appropriate for different settings. Systematic names aren’t usually much good in casual conversation; you can’t do a google search for a sketch of a structural formula; a computer can’t analyze a reaction mechanism using trivial names. It is critical to remember both the human audience and the machine requirements for interpreting and using chemical structure information.
Chemical Structure Drawing Programs (This link is to the Fall 2015 Cheminforamtics OLCC page): http://olcc.ccce.divched.org/2015OLCCModule1P4TLO4 Of notable interest is Dr. Tamas E. Gunda's "Chemical Drawing Programs" (last link on above page): http://www.gunda.hu/dprogs/index.html
EXERCISES
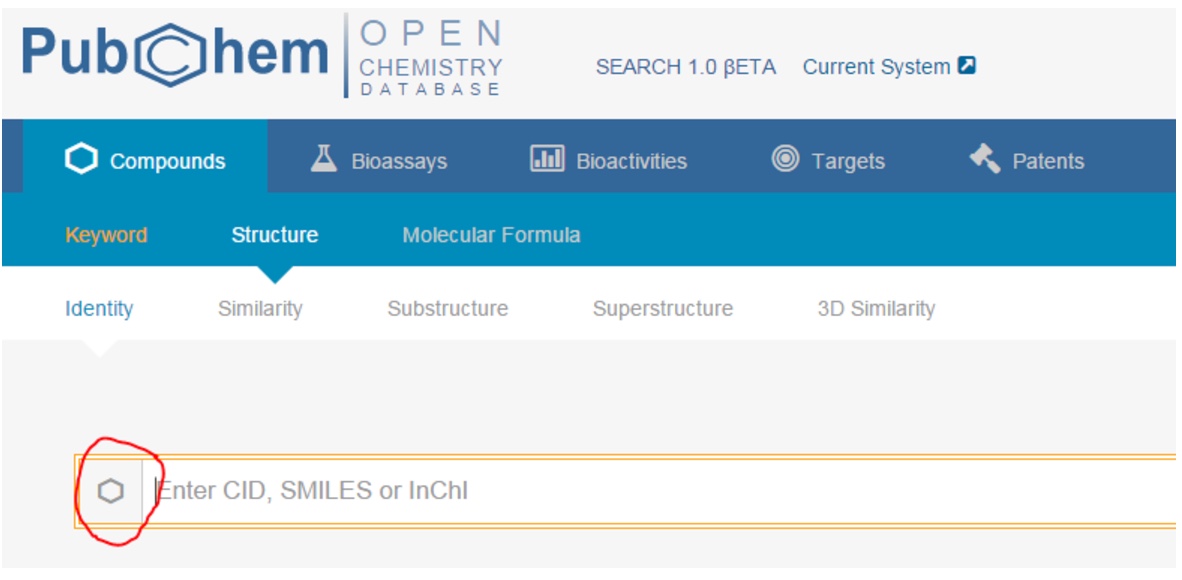
1. Using PubChem’s tool for compound search (go here and click the hexagon to search by structural formula), or other programs of your choice (SciFinder, ChemSpider, Wikipedia (if you dare)), fill in the following table. (For more information, see Exchanging Chemical Representations, above. For more on SMILES and InChI, see the third part of this module.)
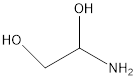
| molecular formula | Structural formula | Systematic name | SMILES | InChI | CAS RN |
|---|---|---|---|---|---|
|
|
CC(=C)C=C | ||||
 |
|||||
|
|
Ammonium acetate | ||||
|
C6H6O |
|||||
|
|
105-53-3 |
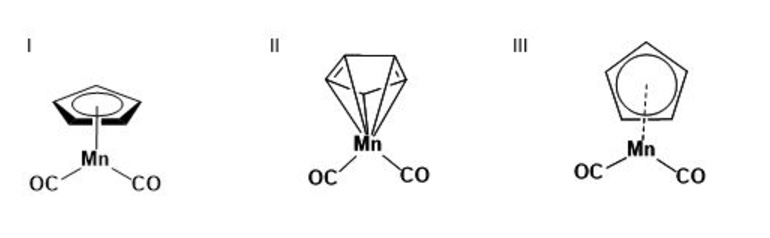
2. Many chemistry databases index by structural formulas based on explicit connectivity for organic small molecules. Many molecules do not fit easily into these conventions for representing bonds, such as coordination compounds and delocalized systems. (Conventions for human-readable and computer-readable graphical representation of such compounds are discussed above.) Of the representations below for a coordination substructure, which is most likely to be acceptable for publication? Which for searching an index? How might each of these representation be interpreted in a database?
3. Resolve each of the following the systematic names listed for Vitamin C into structural formulae using each of the systems below. Is the expected stereochemistry represented? (For more information, see Formulating Chemical Structure Data, above.)
- openmolecules: http://www.openmolecules.org/name2structure
- OPSIN: http://opsin.ch.cam.ac.uk/
- CACTUS: http://cactus.nci.nih.gov/chemical/structure
- ChemSpider: http://www.chemspider.com/
- PubChem: https://pubchem.ncbi.nlm.nih.gov/
- (R)-3,4-dihydroxy-5-((S)-1,2-dihydroxyethyl)furan-2(5H)-one
- (R)-5-((S)-1,2-dihydroxyethyl)-3,4-dihydroxyfuran-2(5H)-one
- (2R)-2-[(1S)-1,2-dihydroxyethyl]-3,4-dihydroxy-2H-furan-5-one
- (5R)-[(1S)-1,2-dihydroxyethyl]-3,4-dihydroxy-3-oxolen-2-one
FURTHER READING & REFERENCES
Formulas
- Jonathan Brecher, “Graphical Representation Standards for Chemical Structure Diagrams (IUPAC Recommendations 2008),” Pure and Applied Chemistry 80, no. 2 (January 1, 2008), 227–410. URL: http://dx.doi.org/10.1351/pac200880020277 (accessed Jan. 2017).
- Antony Williams, “Chemical Structures,” in The ACS Style Guide (American Chemical Society, 2006), 375–83. URL: http://dx.doi.org/10.1021/bk-2006-STYG.ch017 (accessed Sept. 2015).
- Neil G. Connelly and Ture Damhus, eds., IUPAC Nomenclature of Inorganic Chemistry (Cambridge: Royal Society of Chemistry, 2005), 53–67. (The “Red Book”). URL: http://old.iupac.org/publications/books/rbook/Red_Book_2005.pdf (accessed Sept. 2015).
- Wikipedia entry on the Red Book. URL: https://en.wikipedia.org/wiki/IUPAC_nomenclature_of_inorganic_chemistry_2005 (accessed Sept. 2015).
- Compound Interest, http://www.compoundchem.com/ (accessed Sept. 2015).
- (good examples of effective communication using formulas)
Names
ACS/CAS
- “Names and Numbers for Chemical Compounds,” in The ACS Style Guide (American Chemical Society, 2006), 233–54. URL: http://dx.doi.org/10.1021/bk-2006-STYG.ch012 (accessed Sept. 2015).
- American Chemical Society, Naming and Indexing of Chemical Substances for Chemical Abstracts, 2007 Edition (Columbus, OH: American Chemical Society, 2008). URL: http://www.cas.org/File%20Library/Training/STN/User%20Docs/indexguideapp.pdf (accessed Sept 2015).
- IUPAC
- Henri A. Favre and Warren H. Powell, eds., Nomenclature of Organic Chemistry: IUPAC Recommendations and Preferred Names 2013 (Cambridge: Royal Society of Chemistry, 2014). (The “Blue Book”). URL: http://pubs.rsc.org/en/content/ebook/9780854041824 (accessed Sept. 2015).
- Wikipedia entry on the Blue Book. URL: https://en.wikipedia.org/wiki/IUPAC_nomenclature_of_organic_chemistry (accessed Sept. 2015).
- Neil G. Connelly and Ture Damhus, eds., IUPAC Nomenclature of Inorganic Chemistry (Cambridge: Royal Society of Chemistry, 2005), 53–67. (The “Red Book”). URL: http://old.iupac.org/publications/books/rbook/Red_Book_2005.pdf (accessed Sept. 2015).
- Wikipedia entry on the Red Book. URL: https://en.wikipedia.org/wiki/IUPAC_nomenclature_of_inorganic_chemistry_2005 (accessed Sept. 2015).
Cheminformatics
- Blue Obelisk: https://en.wikipedia.org/wiki/Blue_Obelisk
- Warr, W. A. Representation of chemical structures. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 557–579; DOI: 10.1002/wcms.36 (accessed May 29, 2104).
- Warr, W. A. Some Trends in Chem(o)informatics. Chemoinformatics and computational chemical biology. Methods Mol. Biol. 2011, 672, 1–37; DOI: 10.1007/978-1-60761-839-3_1 (accessed May 29, 2104).
- Wild, D. Introducing Cheminformatics: Navigating the world of chemical data. http://i571.wikispaces.com (accessed Sept. 29, 2015).
- Willet, P. Chemoinformatics: a history. WIREs Comput. Mol. Sci. 2011, 1, 46–56; DOI: 10.1002/wcms.1 (accessed May 29, 2014).
Contributors:
- Evan Hepler-Smith, Harvard University
- Leah R. McEwen, Cornell University
- Acknowledgements: Alex Clark, Sunghwan Kim