
6.2: Nucleophilic Acyl Substitution Reactions of Carboxylic Acids
- Page ID
- 469396
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The poor leaving group ability of -OH makes carboxylic acids relatively unreactive towards nucleophilic acyl substitutions. The addition of a strong acid protonates the carbonyl oxygen making the carbonyl carbon more electrophilic and circumventing this problem. Also, the substitution reaction can be promoted by converting -OH into a better leaving group. Despite the lack of reactivity, under the right conditions carboxylic acids can be successfully converted into acid chlorides, acid anhydrides, esters, and amides through nucleophilic acyl substitution.
Conversion of Carboxylic acids to Acid Chlorides
Carboxylic acids can be converted to acid chlorides by reaction with thionyl chloride (SOCl2). During the reaction with thionyl chloride, the hydroxyl group of the carboxylic acid is converted to an acyl chlorosulfite moiety which is a better leaving group. During the reaction a nucleophilic chloride anion is produced which reacts with the acyl chlorosulfite intermediate through nucleophilic acyl substitution to produce an acid halide.
General Reaction

Example

Mechanism
The O=S-Cl bonds of thionyl chloride are analogous to the O=C-Cl bonds of an acid halide in that they can undergo a type of nuleophilicy acyl substitution. During the first step of the mechanism, the carboxylic acid reactant acts as a nucleophile and attacks the electrophilic sulfur of thionyl chloride, pushing the pi electrons of the S=O bond onto oxygen. Reforming the S=O bond and eliminating a cloride anion (Cl-) as a leaving group creates a protonated acyl chlorosulfite. The chlorosulfite group represents an excellent leaving group due to its ability to stabilize a negative charge. The carbonyl of the protonated acyl chlorosulfite intermediate is activated to nucleophilic attack, and promptly reacts with the nucleophilic chloride anion created during the second step of the mechanism to produce a tetrahedral alkoide intermediate. Subsequent reforming of the protonated carbonyl bond eliminates the chlorosulfite group as sulfurdioxide (SO2) and a chloride anion. Lastly, the chloride anion deprotonates the carbonyl to form the acid chloride product.
1) Nucleophilic attack on S=O bond

2) Removal of Cl leaving group

3) Nucleophilic attack on the carbonyl

4) Leaving group removal

5) Deprotonation

Acid Anhydride Formation
An acid anhydride (or just anhydride) is the product of condensation of two carboxylic acid molecules with the release of a water molecule. The most common anhydride in organic chemistry is acetic anhydride, due to the high temperatures needed to remove water.
General Reaction
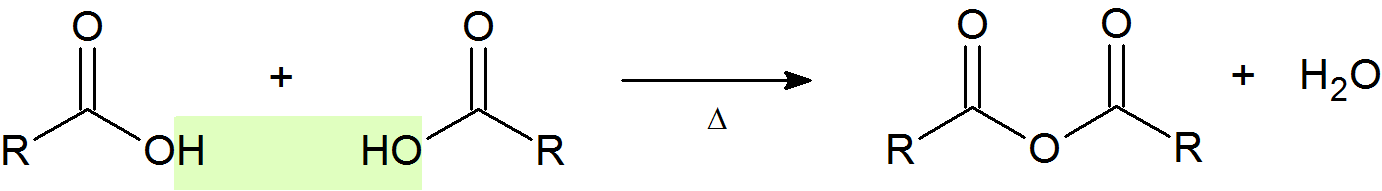
Example

Conversion of Carboxylic Acids into Esters by Alkylation
Carboxylic acids can be easily converted into their conjugate bases through deprotonation with a base, such as sodium hydroxide. The resulting carboxylate can be alkylated using by an SN2 reaction with either a methyl or primary halide. If a methyl ester is required, methyl iodide (CH3I) is a commonly used reagent.
Example

Conversion of Carboxylic Acids to Esters: The Fischer Esterification
Alcohols can be used as nucleophiles to convert carboxylic acids to esters. Due to the poor leaving group ability of -OH in carboxylic acids, an acid catalyst is required to speed up the reaction. Usually the alcohol starting material is used as the reaction solvent so that it can be present in a large excess. This helps to increase the reaction's yield by pushing reaction equilibrium to the right which can be understood via Le Chatelier's principle.
General Reaction

Predicting the Products of a Fischer Esterification

Example

Mechanism
The mechanism begins with protonation of the carbonyl to increase its electrophilic character. A tetrahedral alkoxide intermediate is formed when the alcohol nucleophile adds to the protonated carbonyl, pushing the carbonyl pi electrons onto the oxygen. A different tetrahedral alkoxide intermediate is created when a proton is then transferred to one of the hydroxides (OH), turning it into a good leaving group. Reforming the protonated carbonyl bond then eliminates water as a leaving group. Finally, deprotonation of the carbonyl by water creates the ester product and regenerates the acid catalyst. All the steps of the mechanism are reversible
1) Protonation of the carbonyl

2) Nucleophilic attack on the carbonyl

3) Proton transfer

4) Removal of water as a leaving group

5) Deprotonation

Isotopic Labeling
Evidence to support the Fischer esterification mechanism comes from isotopic labeling experiments with oxygen-18. If the reaction is carried out with an oxygen-18 labeled alcohol, the isotope is found exclusively in the ester and not the water generated. This shows that the C-OH bond of the carboxylic acid and the H-OR bond of the alcohol that is broken during the reaction.

Worked Example: Planning a Synthesis Using a Fischer Esterification
How could the following molecule be made using a Fischer Esterification?

Analysis

The key bond formed during a Fischer Esterification is the C-O sigma bond. The target molecule can be separated into the two required starting materials by breaking this bond. The carbonyl carbon fragment gains an –OH to become a carboxylic acid and the oxygen fragment gains an H to become an alcohol. The carboxylic acid and the alcohol are reacted using a strong acid catalyst.
Solution

Direct Conversion of Carboxylic Acids to Amides
The direct reaction of a carboxylic acid with an amine would be expected to be difficult because the basic amine would deprotonate the carboxylic acid to form a stable carboxylate salt. However when the ammonium carboxylate salt is heated to a temperature above 100 oC, water is driven off and an amide is formed. Due to the extreme conditions, this reaction is rarely used. When converting to an amide it is preferred to convert the carboxylic acid to a more reactive form such as an acid chloride first and then convert this molecule to an amide.
General Reaction

Conversion of Carboxylic Acids to Amides using DCC
The formation of amides from carboxylic acids and amines is a reaction of great importance in biochemistry. Proteins are formed through the creating of amide bonds between amino acid residues, so a great deal of research has been performed to find efficient methods. The direct conversion of a carboxylic acids to amides is difficult because amines are basic and tend to convert carboxylic acids to their highly unreactive carboxylates. One solution to this problem is through the use of dialkylcarbodiimides (R-N=C=N-R) coupling reagents, such as dicyclohexylcarbodiimide (DCC).

During a DCC amide coupling, the OH of a carboxylic acid is made into a good leaving group which can then be replaced by an amine during nucleophilic acyl substitution. Using DCC as a coupling reagent, 1o and 2o amines can be used to create 2o and 3o amides respectfully.
Basic reaction

Predicting the products of DCC Coupling

Example

Mechanism
DCC deprotonates the carboxylic acid to form a carboxylate, which is a better nucleophile. The corresponding protonation of DCC increases the electrophilic character of its C=N imide bond. The caboxylate nucleophile then adds to the imide bond and pushes the C=N pi electrons onto the nitrogen. The amine nucleophile can now add itself to the carbonyl bond as part of a nuleophilic acyl substitution. The DCC coupled oxygen is eliminated as dicyclohexylurea, a good leaving group, to create the amide product.
1) Deprotonation

2) Nucleophilic attack by the carboxylate

3) Nucleophilic attack by the amine

4) Proton transfer

5) Leaving group removal

Conversion of Carboxylic acids to 1o alcohols
Lithium aluminum hydride (LiAlH4)
Hydride nucleophiles from lithium aluminum hydride (LiAlH4) can reduce carboxylic acids to 1o alcohols. Note that NaBH4 is not a strong enough reducing agent to convert carboxylic acids or esters to alcohols. Because the incoming nucleophile is an “H” the reaction first produces an aldehyde intermediate which available for further hydride additions. The aldehyde intermediate is difficult to isolate because it is more reactive than the original carboxylic acid. This reaction represents the first example in this chapter where a carboxylic acid derivative can undergo a double nucleophilic addition. In the mechanism of this reaction, a nucleophilic acyl substitution is followed by a nucleophilic addition allowing for two hydride nucleophiles being added to the electrophilic carbonyl carbon of a carboxylic acid.
General reaction

Predicting the product of a hydride reduction

Example

Possible Mechanism
Although the mechanism of this reaction is not precisely known, much of it is understood. Initially, a hydride deprotonates the carboxylic acid to form a lithium carboxylate, hydrogen gas (H2), and aluminum hydride. Then a hydride nucleophile, from aluminum hydride, adds to the carbonyl carbon as part of a nucleophile acyl substitution. The resulting high-energy dianion intermediate forms a Lewis Acid/Base complex with aluminum, making one of the oxygens a good leaving group (the negative charge on the oxygen complexed to aluminum is not shown in step 2 below). The carbonyl bond is reformed along with the elimination of OAlH2 as a leaving group to form an aldehyde. A hydride nucleophile from another molecule of LiAlH4 adds to the re-formed carbonyl carbon as part of a nucleophilic addition. The resulting alkoxide intermediate is protonated during an acidic work-up to form the 1o alcohol product. Due to the formation of a dianion intermediate the reaction requires relatively high temperature and long reaction times.
1) Deprotonation

2) Nucleopilic attack by a hydride anion

3) Leaving group removal

4) Nucleophilic attack by a hydride anion

5) Alkoxide protonation
