
15.1: Introduction to Substitution Reactions
- Page ID
- 282015

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)
Chemical Reactivity and Reactions of Complexes
In this chapter we will discuss the chemical reactivity and reactions of coordination compounds. First, let us briefly review what are the basic parameters that define if a reaction can be observed or not. The first parameter is the Gibbs free energy of reaction ΔG. The Gibbs free energy defines the thermodynamic driving force for the reaction. The more negative ΔG is, the greater the thermodynamic driving force. The Gibbs free energy of reaction is a measure for the difference in the stability of the reactants and products. The less stable the reactants, and the more stable the products, the more negative ΔG. The second parameter is the kinetics of a reaction. The kinetics is a measure of the speed at which a reaction occurs. The kinetics is independent from the thermodynamics. This means that even if there is a strong thermodynamic driving force for the reaction, the rate of the reaction may be extremely slow, and practically no reaction is observed. For example, graphite is thermodynamically more stable than diamond at ambient pressure and temperature, but the kinetics of the conversion at room temperature is so slow, so that practically no reaction is observed. The rate of the reaction is determined by the the activation energy that needs to be overcome to make the reaction happen. The lower the activation energy, the faster the reaction. In coordination chemistry the activation energy and the reaction rates are associated with the inertness and the lability of complexes. The more inert and the less labile a complex is the higher the activation barrier and the more slowly it reacts. The lability is the opposite of the inertness and is defined by the half-life time of a dative bond. The greater the half-life time, the more inert, and the less labile the complex.
Inert and Labile Complexes
The greater the half-life time of a dative bond, the more inert, and the less labile the complex. Where do we actually draw the borderline between a labile and an inert complex? A commonly accepted definition is that complexes with metal-ligand bond half life times of <30 s at standard conditions are considered labile complexes with a low activation energy. Those complexes undergo fast substitution reactions, and the chemical equilibrium is reached fast (Fig. 9.1.1).
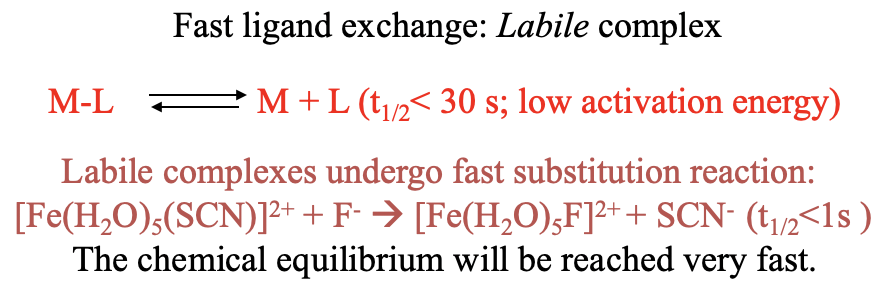
An example is the substitution of a thiocyanato ligand in a pentaaquathiocyanato iron (2+) complex by a fluoro ligand. This substitution reaction is fast because the half-life time of the metal-ligand bond is smaller than a second.
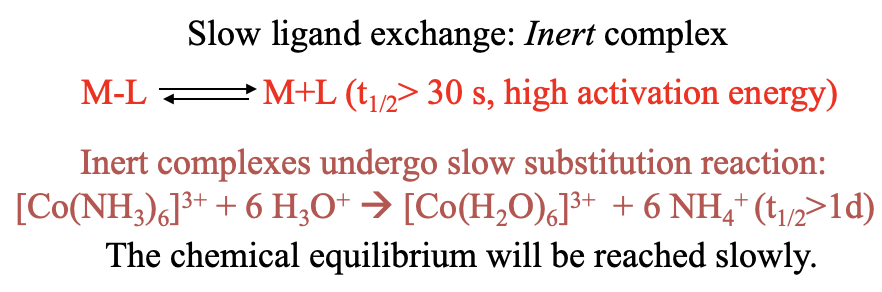
If the half-life time is greater than 30s, then the complex is considered inert. For example, the hydrolysis of hexaammine cobalt (3+) is slow because the half-life time of the Co-N bond is larger than a day. The chemical equilibrium is reached only slowly (Fig. 9.1.2).
Can we predict which complexes are labile, and which ones are inert? Many factors determine this, but there are some trends we can keep in mind. For transition metals, octahedral d3, low spin d4-d6, and square planar d8 complexes tend to be inert. A second trend is that the lability decreases with the period within a group. For instance, we would expect that in group 10 the lability decreases from Ni to Pd to Pt. This is because covalency of the bonds tends to increase with increasing period due to the softer nature of higher period metals. A third trend is that higher positive charges at the metal ion lead to a decrease in liability. Metal ions with higher oxidation numbers also tend to make more covalent bonds because the high positive charge at the metal is stabilized by covalent interactions (Fig. 9.1.3).

We can also compare the d-block metals with the other blocks. S-block metal complexes tend to have the greatest lability, followed by the f-block, followed by the d-block. P-block metal complexes can widely vary in lability (Fig. 9.1.4).

Also the nature of the ligand influences the lability. Generally, when the ligand is a good leaving group, then the bond tends to be labile. A ligand is usually a good leaving group when it is stable in the uncoordinated form. For instance, a chloro ligand is a good leaving group because chloride anions are quite stable in their non-coordinated form. An alkyl ligand on the other hand would not be considered as a good leaving group because the an alkylide anion is not a very stable species in the free form.
Stability of Complexes
Now let us have a closer look at the stability of complexes. The more negative the Gibbs free energy of formation, the greater the equilibrium constant of formation K. The larger the equilibrium constant K of formation, the more stable the complex. This can be expressed by the van’t Hoff equation ΔG = -RTlnK.
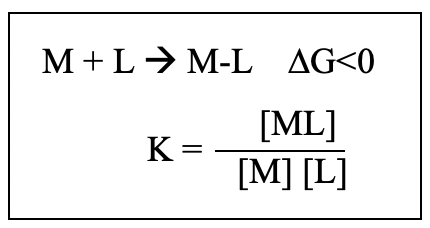
For the formation of a single dative bond M-L the constant of formation is given by K=[ML]/[M][L] (Fig. 9.1.5). The greater K, the farther the equilibrium is on the side of the product. K is usually reported as logK because K can vary over 35 orders of magnitude.
Rationalizing the Different Stability of Complexes
How can we rationalize the different stabilities of coordination bonds, and coordination compounds? Here, the HSAB concept is very useful. Hard-hard and soft-soft interactions are typically strong and lead to high constants of formation and stable complexes. Hard-soft interactions lead to small constants of formation and less stable complexes.

Let us understand this better by the example of the interaction of Ag+ with different ligands (Fig. 9.1.6). You can see in the table the logK values for the different ligands. Br- has the largest one, followed by NH3 and Cl-. F- has the smallest one. Can we explain this with the HSAB concept? Ag+ is a soft cation, thus it would be expected to make the strongest interactions with the softest ligand. Bromide is the softest of the four, and hence HSAB correctly predicts the stability. F- is the hardest ligand and would be expected to make the weakest complex. This is in accordance with the small logK value for F-. NH3 and Cl- have similar intermediate hardness, which is consistent with the similar, intermediate logK values.
Stabilities of Multi-ligand Complexes

How does the stability of a complex change as more ligands are added? Let us take a look at the constants of formation to answer the question. The addition of an additional ligand is associated with an additional chemical equilibrium which relates to an additional constant of formation. The number of equilibrium constants is equal to the number of ligands. For instance, when we have a complex ML4 with four ligands, then for the addition of the first ligand we get a constant K1= [ML] / [M] [L] , for the second ligand we get a constant K2=[ML2] / [ML] [L] , for the third ligand there is a constant K3=[ML3] / [ML2] [L], and for the fourth ligand there is a constant K4=[ML4] / [ML3] [L]. When we analyze the K-values, we notice that they decline from K1 to K4. For a general complex MLn we find that the constants decline from K1 to Kn. How can this be explained? The explanation is that the more ligands are getting added, the less many free ligands are available that could be further added. Thus, the probability that further ligands are added decreases, and this results in a smaller equilibrium constant. So it is a statistical effect that explains the phenomenon.
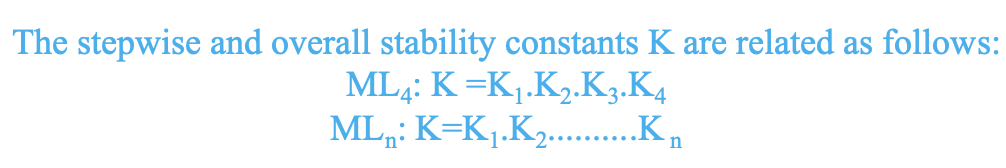
Then what is the overall stability constant K of the complex associated with the equation M + 4 L → ML4 and more general M + nL → MLn? It is the product of the constants associated with each step. For the complex ML4 it is K=K1K2K3K4. For the general complex MLn it is K=K1K2.........Kn (Fig. 9.1.8).
There are exceptions from the rule that the stability constants of complexes associated with the addition of one ligand decreases with the number of ligands. If there is an exception, then this is because the ligand addition causes a major change in the electronic structure of the complex. Here are a couple of examples.
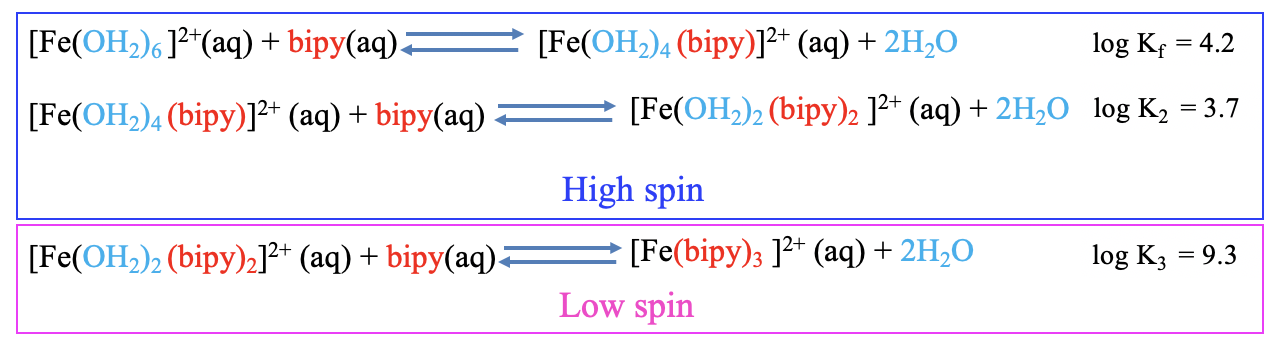
In the first example (Fig. 9.1.9) the aqua ligands of a hexaaqua iron (2+) complex are substituted by bipyridyl ligands. The bipyridyl ligand is a bidentate ligand, and so one bipydridyl ligand substitutes two aqua ligands. In the first substitution, the two aqua ligands get replaced by one bipyridyl ligand to form a tetraaqua(bipyridyl) iron (2+) complex. This step is associated with a logK1 value of 4.2.
In the second substitution step, two aqua ligands of the tetraaqua(bipyridyl) iron (2+) complex get replaced by another bipyridyl ligand to form a diaqua bis(bipyridyl) iron(2+) complex. As expected this step is associated with a smaller logK2 value of 3.7.
In the third step, the last two aqua ligands get substituted by another bipyridyl ligand forming a tris(bipyridyl) iron(2+) complex. What is the logK3 value? It is 9.3! This is much larger than logK1 and logK2, so this result is unexpected. A major change in electronic structure must have occurred. What is it? In this case it is a change from a high-spin to a low spin complex. The bipyridyl ligands are strong π-acceptors that increase Δo. Substitution of the first four aqua ligands increases ΔO but the increase is not large enough yet so that it induces a switch from a high-spin to a low-spin complex. Only the third bipyridyl ligand does that. As a consequence, the formation constant logK3 is unusually large.
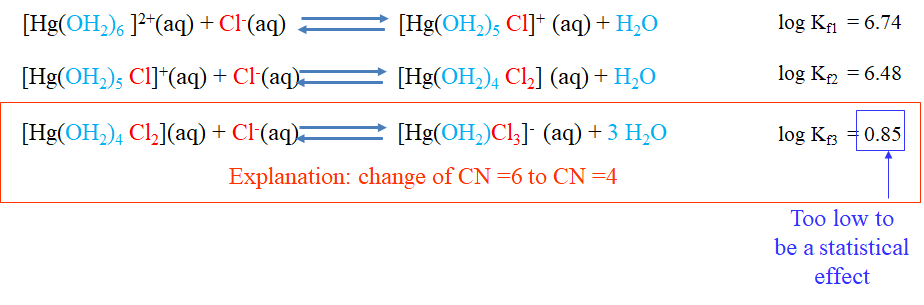
In the second example (Fig. 9.1.10) we subsequently substitute aqua ligands by chloroligands in a hexaaqua mercury(2+) complex. The first substitution leads to a pentaaquachloro mercury(+) complex. This reaction is associated with a logK1 value of 6.74. The second substitution leads to a tetraaquadichloro mercury(0) complex, and this reaction is associated with a smaller logK2 value of 6.48. The third substitution reaction leads to an aquatrichloro mercury(1-) complex. The logK3 value for this reaction is 0.85. We see that this value is smaller than logK2, and this is what we expected, but the difference between logK3 and logK2 is much larger than that between logK1 and logK2. The increased difference is too large to be just caused by a statistical effect, but must be induced by a change in electronic structure. In this case it is a change in coordination number. As the third chloro ligand is added, the complex does not lose only one but three aqua ligands. This reduces the coordination number from six to four.
Stability of Chelate Complexes
The Thermodynamic Chelate Effect
Chelating complexes tend to be more stable than complexes with monodentate ligands. This is called the “thermodynamic chelate effect”. The effect deserves an explanation. The explanation is the increase of entropy that occurs when two or more monodentate ligands are replaced by a chelating ligand. The entropy increases because the overall number of particles increases as the substitution takes place.
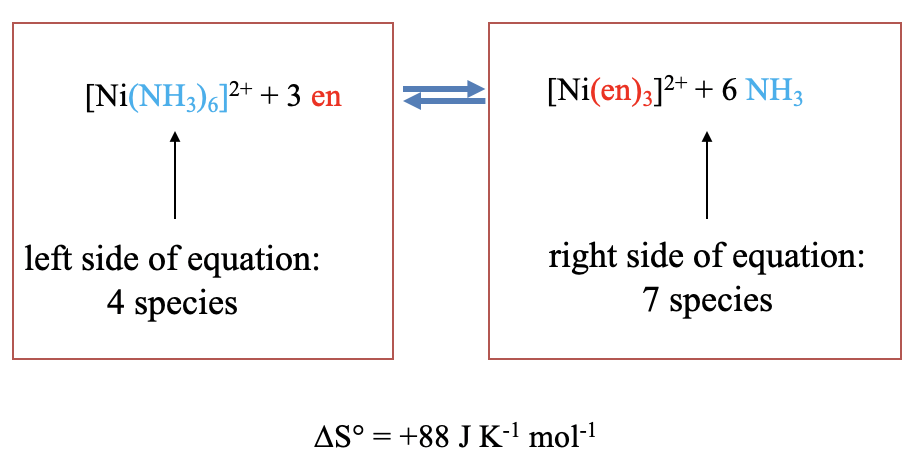
For example, the substitution of six ammine ligands in the hexaammine nickel (2+) complex by three ethylenediamine chelating ligands increases the number of molecules from four to seven, and hence the entropy increases, in this case by 88 J K-1 and mol-1 (Fig. 9.1.11)
The Kinetic Chelate Effect
In addition to the thermodynamic chelate effect, there is the kinetic chelate effect. Chelate complexes are frequently more inert than complexes with monodentate ligands. Chelate complexes are more inert for two reasons (Fig. 9.1.12).
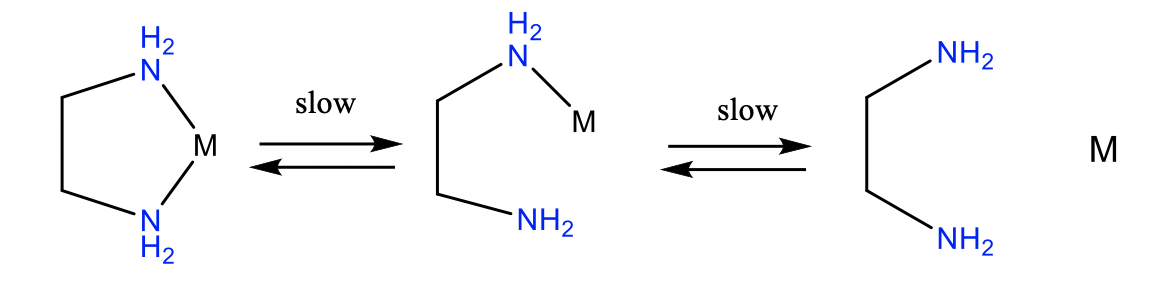
Firstly, the whole ligands needs to rotate and bend in order to cleave the first metal-ligand bond. This requires time and slows the kinetics of the bond cleavage. The second reason is that the detached donor atom cannot leave the proximity of the complex because the ligand is still attached via the other donor atom. This increases the probability of the re-formation of the metal-ligand bond which decreases the probability of both bonds being cleaved.
Mechanism of Ligand Substitution Reactions
Now let us talk about the mechanism of ligand substitution reactions. We know two basic mechanisms according to which a ligand substitution can occur. One is called the intimate mechanism, and the other one is called the stoichiometric mechanism.
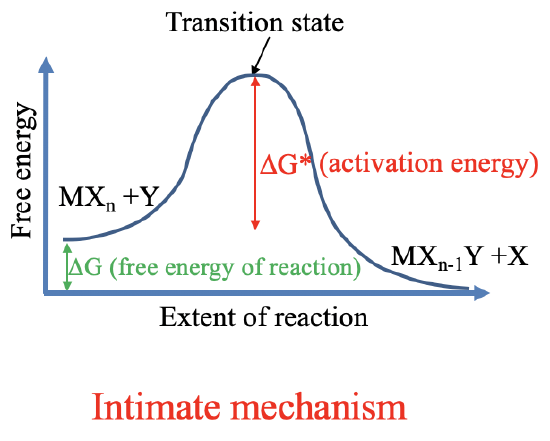
The intimate mechanism goes through a single transition state as the reaction proceeds.
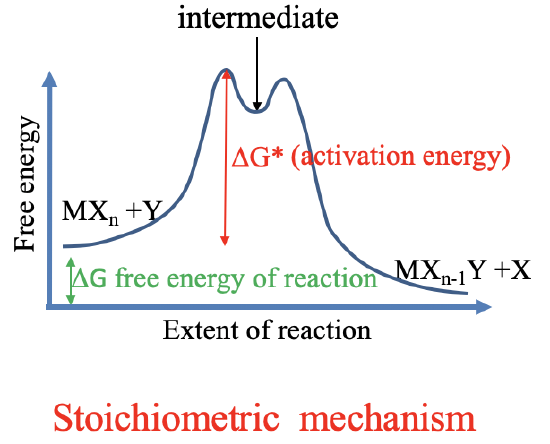
The stoichiometric mechanism goes through an intermediate. The intermediate is a shallow local minimum on the reaction coordinate. The transition state is the maximum on the reaction coordinate. For both mechanisms the principle of microscopic reversibility holds. This means that the reverse reaction follows the same free energy path as the forward reaction. For both mechanisms the free Gibbs energy of reaction is given by the difference between the free energy of the products and the free energy of the reactants. The reaction can only spontaneously occur if the free energy of the products is smaller than the free energy of the reactants. The activation energy is the free energy difference between the transition state and the reactants.
Classifications of Mechanisms
There are two forms of the stoichiometric mechanism: The dissociate mechanism D and the associative mechanism A. In the dissociative mechanism (Fig. 9.1.15), an old ligand X is lost first, and the intermediate has a lower coordination number. In the associative mechanism (Fig. 9.1.16), the new ligand Y is added first and the intermediate has a higher coordination number. The intermediates must be detectable, thus must be present in relatively high concentration to measurable with an analytical technique. This requires are relatively deep local thermodynamic minimum for the intermediate. In practice, many local minima are too shallow to allow for the clear detection of the intermediate. We call such a mechanism an Interchange mechanism. There are two forms of interchange mechanisms: Ia and Id. Ia stands for associative interchange mechanism and ID stands for dissociative interchange mechanism. Overall we can have a continuous range of mechanisms that range from A to Ia to intimate to Id to D.
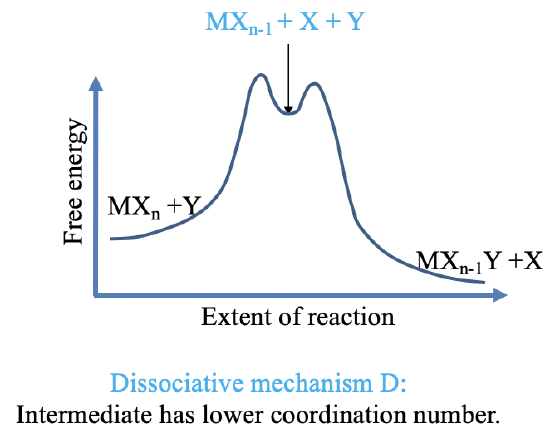
For a dissociative and the Id mechanism the dissociation is the rate limiting step. This is because the activation energy is associated with the dissociative step. We need to go strongly uphill to achieve dissociation, and go into the local thermodynamic minimum. From, there we only need to go slightly uphill to add the new ligand.
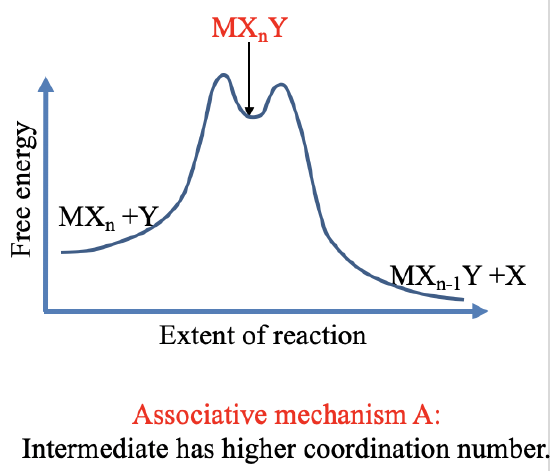
For the associative and the Ia mechanism (Fig. 9.1.16), the associative step is the rate limiting step because the activation energy is associated with the associative step. In this case we first need to go energetically strongly uphill to achieve association and reach the local thermodynamic minimum. We only need to go slightly uphill from the local thermodynamic minimum to lose the old ligand.
How to Distinguish Between D/Id and A/Ad
We can use the relationships between the rate determining step and the mechanism to experimentally distinguish between associative A/Ia and dissociative D/Id mechanisms. If the reaction rate strongly depends on the new ligand, then we likely have an associative or Ia mechanism. This is because the associative step is the rate determining step. If the reaction rate does not strongly depend on the new ligand, then the mechanism is likely dissociative D or Id. This is because in the dissociate mechanism, the addition of the new ligand is not the rate-determining step.
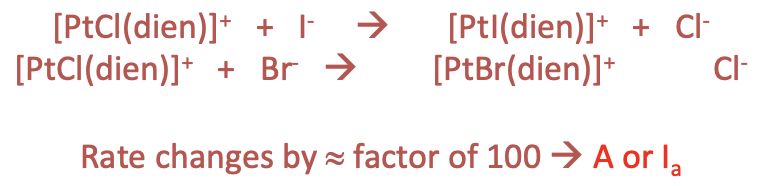
For example, we can substitute a chloro ligand in a [PtCl(dien)]+ complex by iodo and bromo ligands, respectively (Fig. 9.1.17). What we measure is that the reaction rates are by a factor of 100 different. This means that the reaction rates does depend on the new ligand and this indicates an associative or Ia mechanism.
Steric arguments can help to predict if a mechanism is associative or dissociative. Lower coordination numbers and little steric crowding tends to favor A and Ia mechanisms, while higher coordination numbers and greater steric crowding tend to favor the dissociative mechanism.
Dr. Kai Landskron (Lehigh University). If you like this textbook, please consider to make a donation to support the author's research at Lehigh University: Click Here to Donate.