
8.5: Elimination reactions
- Page ID
- 225806
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Alkyl halides undergo elimination via two common mechanisms, known as E2 and E1, which show some similarities to SN2 and SN1, respectively. In E2, elimination shows a second order rate law, and occurs in a single concerted step (proton abstraction at Cα occurring at the same time as Cβ-X bond cleavage). In E1, elimination goes via a first order rate law, in two steps (Cβ-X bond cleavage occurring first to form a carbocation intermediate, which is then ‘quenched’ by proton abstraction at the alpha-carbon).
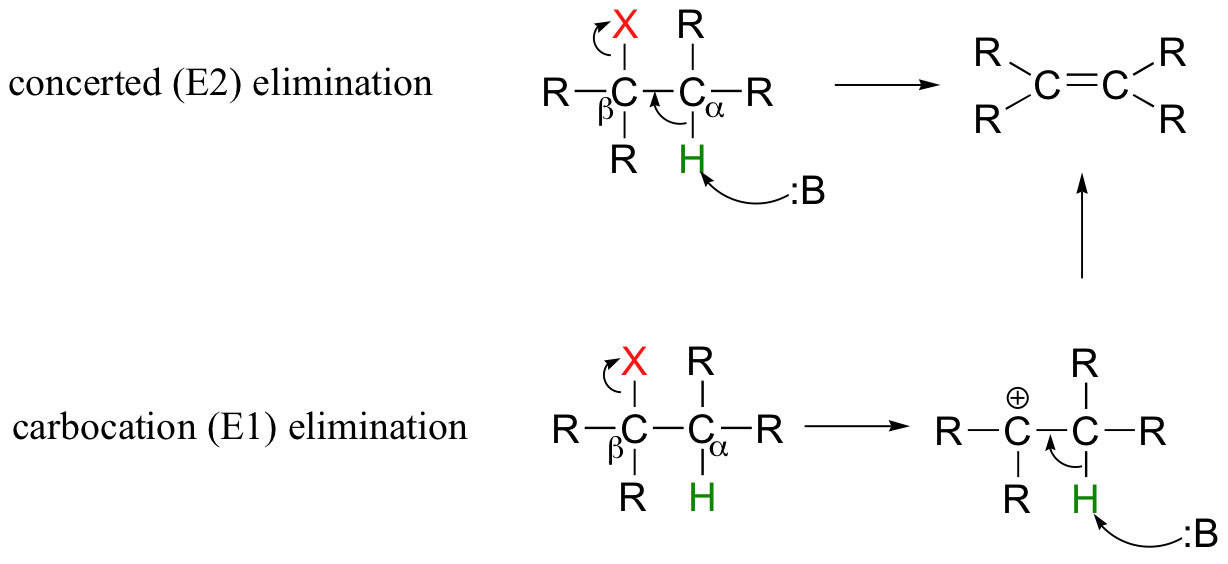 These mechanisms are important in laboratory organic chemistry. As explained below, which mechanism actually occurs in a laboratory reaction will depend on the identity of the R groups (ie., whether the alkyl halide is primary, secondary, tertiary, etc.) as well as on the characteristics of the base.
These mechanisms are important in laboratory organic chemistry. As explained below, which mechanism actually occurs in a laboratory reaction will depend on the identity of the R groups (ie., whether the alkyl halide is primary, secondary, tertiary, etc.) as well as on the characteristics of the base. E1 and E2 reactions in the laboratory
E2 elimination reactions in the laboratory are carried out with relatively strong bases, such as alkoxides (deprotonated alcohols, –OR). 2-Bromopropane will react with ethoxide, for example, to give propene.

Propene is not the only product of this reaction, however – the ethoxide will also to some extent act as a nucleophile in an SN2 reaction.
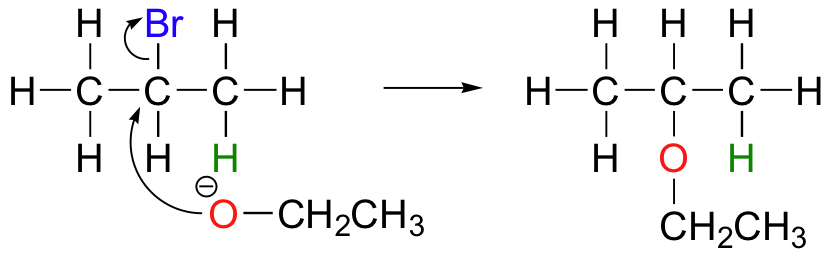
Chemists carrying out laboratory nucleophilic substitution or elimination reactions always have to be aware of the competition between the two mechanisms, because bases can also be nucleophiles, and vice-versa. However, a chemist can tip the scales in one direction or another by carefully choosing reagents. Primary carbon electrophiles like 1-bromopropane, for example, are much more likely to undergo substitution (by the SN2 mechanism) than elimination (by the E2 mechanism) – this is because the electrophilic carbon is unhindered and a good target for a nucleophile.
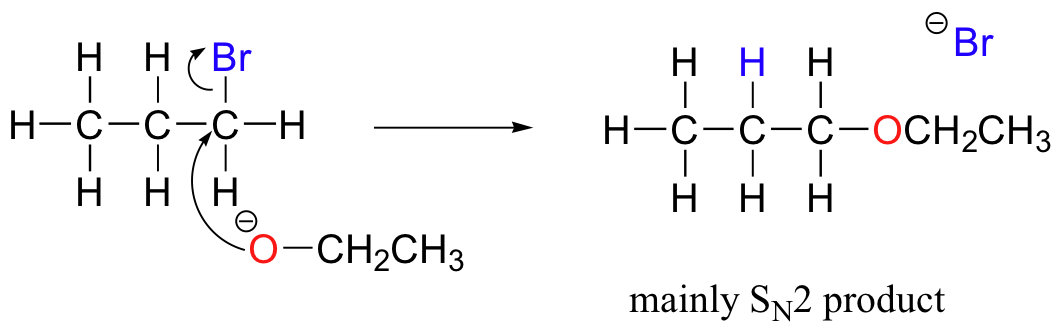
SN1 and E1 mechanisms are unlikely with such compounds because of the relative instability of primary carbocations.
The nature of the electron-rich species is also critical. Acetate, for example, is a weak base but a reasonably good nucleophile, and will react with 2-bromopropane mainly as a nucleophile.
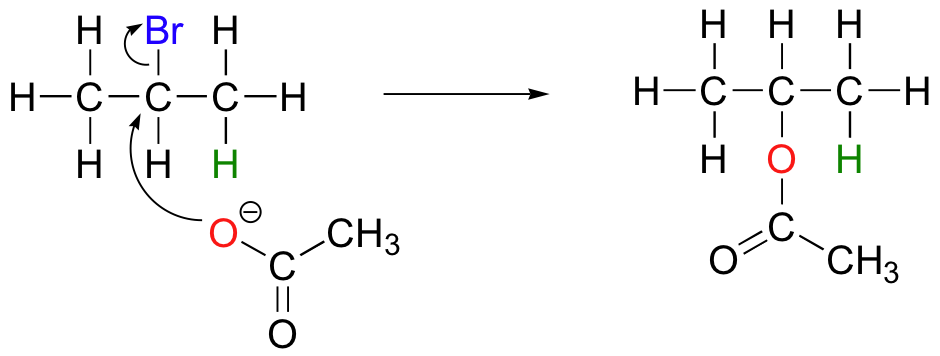
In order to direct the reaction towards elimination rather than substitution, heat is often used. This is because elimination leads to an increase in the number of molecules (from two to three in the above example), and thus an increase in entropy. High temperatures favor reactions of this sort, where there is a large increase in entropy. Substitution does not usually involve a large entropy change, so if SN2 is desired, the reaction should be done at the lowest temperature that allows substitution to occur at a reasonable rate.
Also, a strong hindered base such as tert-butoxide can be used. The bulkiness of tert-butoxide makes it difficult for the oxygen to reach the carbon (in other words, to act as a nucleophile). It is more likely to pluck off a proton, which is much more accessible than the electrophilic carbon).

E1 reactions occur by the same kinds of carbocation-favoring conditions that have already been described for SN1 reactions (section 8.3.): a secondary or tertiary substrate, a protic solvent, and a relatively weak base/nucleophile. In fact, E1 and SN1 reactions generally occur simultaneously, giving a mixture of substitution and elimination products after formation of a common carbocation intermediate. When tert-butyl chloride is stirred in a mixture of ethanol and water, for example, a mixture of SN1 products (2-methylpropan-2-ol and tert-butyl ethyl ether) and E1 product (2-methylpropene) results. Heat is used if elimination is desired, but mixtures are still likely.
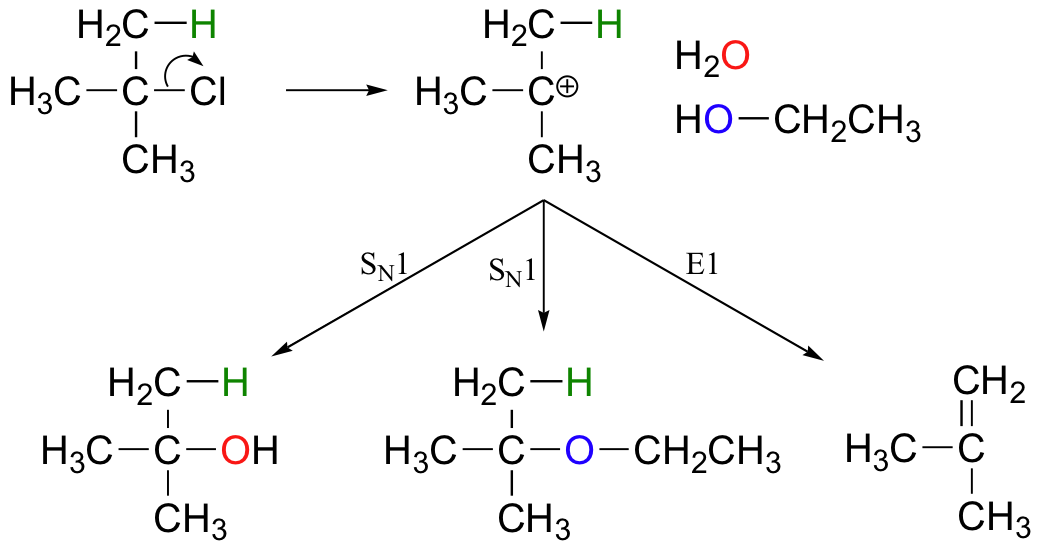
Exercise
[reveal-answer q=”673235″]Show Solution[/reveal-answer]
[hidden-answer a=”673235″]
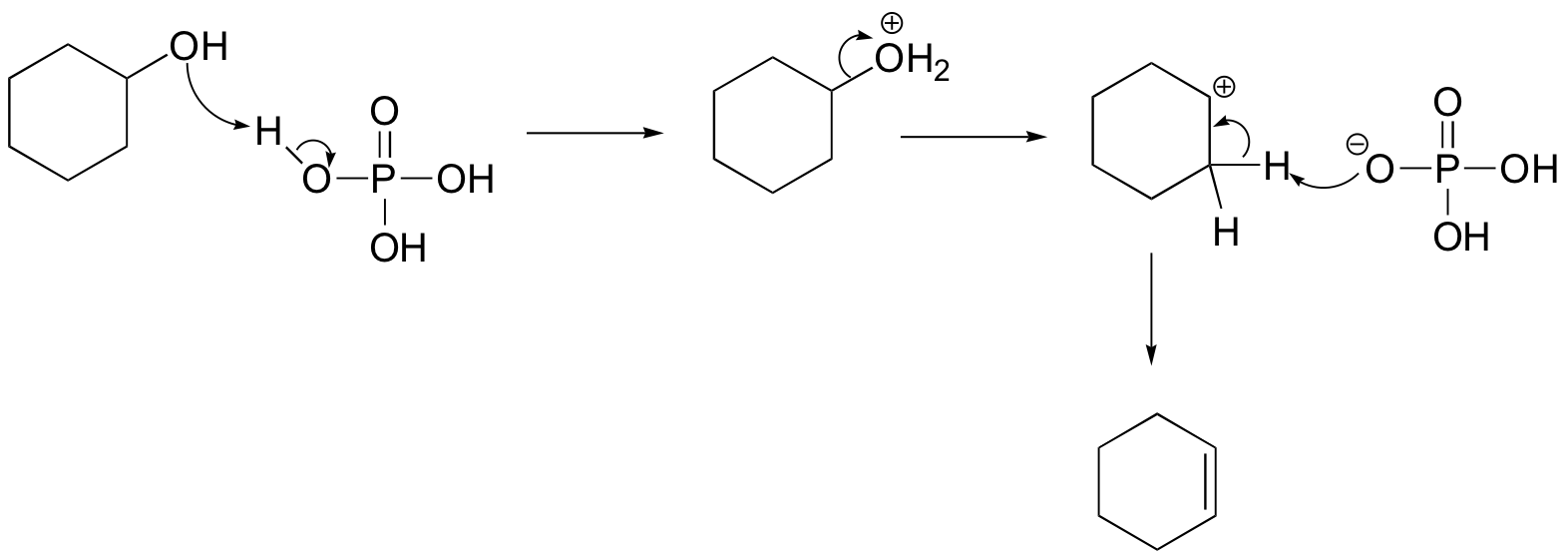
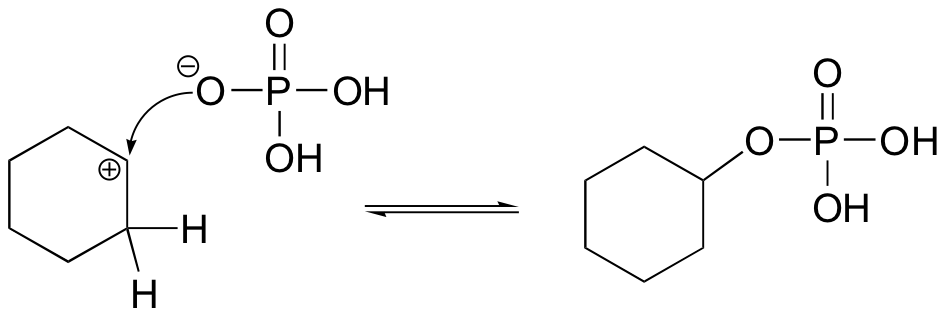
The cyclohexyl phosphate could form if the phosphate attacked the carbocation intermediate as a nucleophile rather than as a base:
[/hidden-answer]
Next, let’s put aside the issue of competition between nucleophilic substitution and elimination, and focus on the regioselectivity of elimination reactions. In many cases an elimination reaction can result in more than one constitutional isomer or stereoisomer. The elimination products of 2-chloropentane provide a good example:

This reaction is both regiospecific and stereospecific. In general, more substituted alkenes are more stable, and as a result, the product mixture will contain less 1-butene than 2-butene (this is the regiochemical aspect of the outcome, and is often referred to as Zaitsev’s rule). In addition, trans–alkenes are generally more stable than cis-alkenes, so we can predict that more of the trans product will form compared to the cis product.
However, certain other eliminations (which we will not be studying) favor the least substituted alkene as the predominant product, due to steric factors. Such a product is known as the Hoffmann product, and it is usually the opposite of the product predicted by Zaitsev’s Rule.
E2 eliminations
E2, bimolecular elimination, was proposed in the 1920s by British chemist Christopher Kelk Ingold. Unlike E1 reactions, E2 reactions remove two substituents with the addition of a strong base, resulting in an alkene.
Introduction
E2 reactions are typically seen with secondary and tertiary alkyl halides, but a hindered base is necessary with a primary halide. The mechanism by which it occurs is a single step concerted reaction with one transition state. The rate at which this mechanism occurs is second order kinetics, and depends on both the base and alkyl halide. A good leaving group is required because it is involved in the rate determining step. The leaving groups must be coplanar in order to form a pi bond; carbons go from sp3 to sp2 hybridization states.
General Reaction
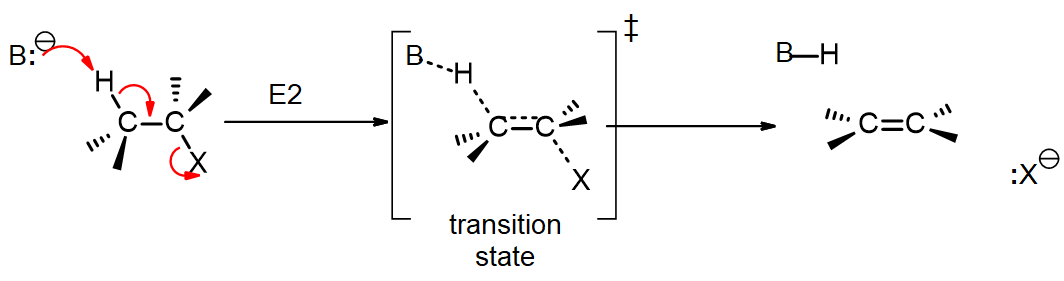
In this reaction B¯ represents the base and X represents a leaving group, typically a halogen. There is one transition state that shows the single step (concerted) reaction. The base is forming a bond to the hydrogen, the pi bond is forming, and the C-X bond is beginning to break. In most reactions this requires everything to be in the same plane, and the leaving group 180o to the H that leaves; the H and the X are said to be “antiperiplanar”.
In summary, An E2 reaction has certain requirements to proceed:
- A strong base is necessary especially necessary for primary alkyl halides. Secondary and tertiary primary halides will procede with E2 in the presence of a base (OH-, RO-, R2N-)
- Both leaving groups (the H and the X) should be on the same plane, this allows the double bond to form in the reaction. In the reaction above you can see both leaving groups are in the plane of the carbons.
- Follows Zaitsev’s rule, the most substituted alkene is usually the major product.
- Hoffman Rule, if a sterically hindered base will result in the least substituted product.
The reaction coordinate free energy diagram for an E2 reaction shows a concerted reaction:
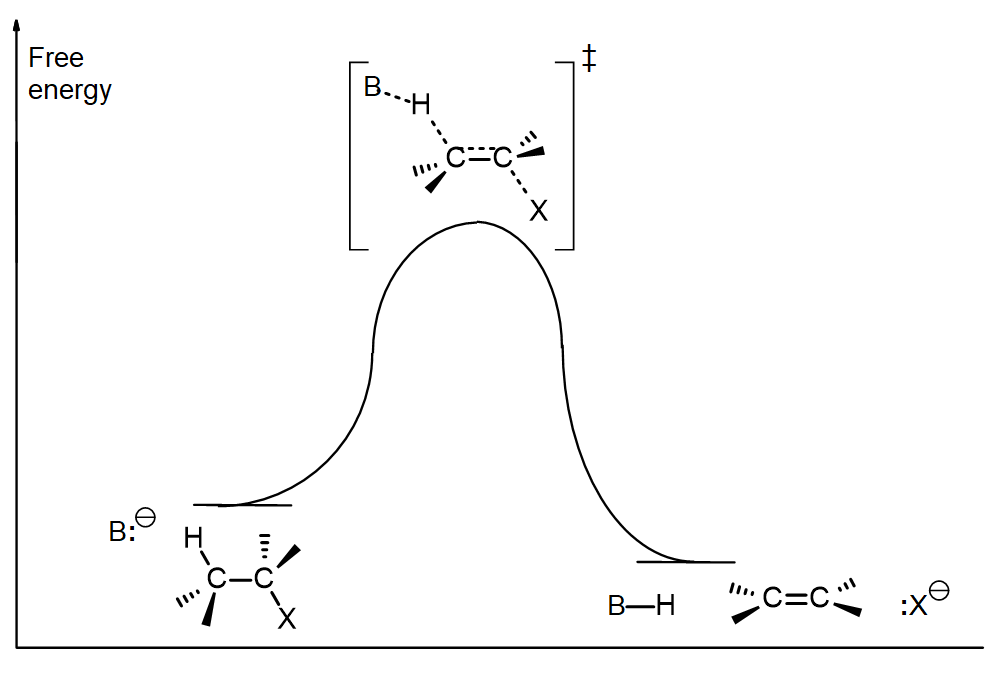
Key features of the E2 elimination
The main features of the E2 elimination are:
- It usually uses a strong base (often –OH or –OR) with an alkyl halide
- Primary, secondary or tertiary alkyl halides are all effective reactants, with tertiary reacting most easily. With primary alkyl halides, a substituted base such as KOtBu and heat are often used to minimize competition from SN2.
- The H and the leaving group should normally be antiperiplanar (180o) to one another.
- Zaitsev’s Rule applies, unless a very hindered base such as KOtBu is used, so the more substituted alkene is usually major.
References
- Vollhardt, K. Peter C., and Neil E. Schore. Organic Chemistry Structure and Function. New York: W. H. Freeman, 2007.
- Organic chemistry, by Marye Anne Fox, James K. Whitesell
E1 Reactions
Unimolecular elimination (E1) is a reaction in which the removal of an HX substituent results in the formation of a double bond. It is similar to a unimolecular nucleophilic substitution reaction (SN1) in particular because the rate determining step involves heterolysis (losing the leaving group) to form a carbocation intermediate. Because the rate determining (slow) step involves only one reactant, the reaction is unimolecular with a first order rate law. Since these two reactions behave similarly, they compete against each other. Many times, both will occur simultaneously to form different products from a single reaction. However, one can be favored over the other by using hot or cold conditions.
General reaction
An E1 reaction involves the deprotonation of a hydrogen nearby (usually one carbon away, or the beta position) the carbocation resulting in the formation of an alkene product. In order to accomplish this, a base is required. For a simplified model, we’ll take B to be a base, and LG to be a halogen leaving group.
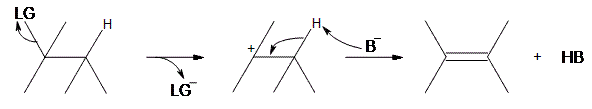
As can be seen above, the preliminary step is the leaving group (LG) leaving on its own. Because it takes the electrons in the bond along with it, the carbon that was attached to it loses its electron, making it a carbocation. Once it becomes a carbocation, a base ($$B^-$$) deprotonates the intermediate carbocation at the beta position, which then donates its electrons to the neighboring C-C bond, forming a double bond. Unlike E2 reactions, which require the proton to be anti to the leaving group, E1 reactions only require a neighboring hydrogen. This is due to the fact that the leaving group has already left the molecule. The final product is an alkene along with the HB byproduct.
Reactivity
Due to the fact that E1 reactions create a carbocation intermediate, rules present in $$S_N1$$ reactions still apply.
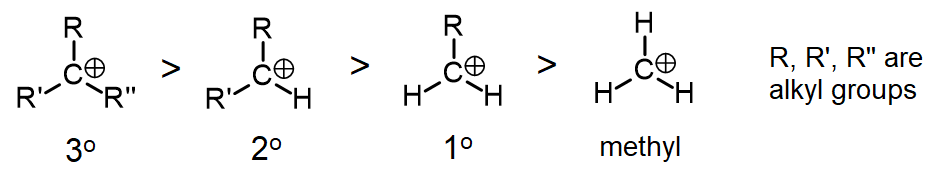
As expected, tertiary carbocations are favored over secondary, primary and methyls. In general, primary and methyl carbocations do not proceed through the E1 pathway for this reason, unless there is a means of carbocation rearrangement to move the positive charge to a nearby carbon. Secondary and tertiary carbons form more stable carbocations, thus this formation occurs quite rapidly.
Secondary carbocations can be subject to the E2 reaction pathway, but this generally occurs in the presence of a good / strong base. Adding a weak base to the reaction disfavors E2, essentially pushing towards the E1 pathway. In many instances, solvolysis occurs rather than using a base to deprotonate. This means heat is added to the solution, and the solvent itself deprotonates a hydrogen. The medium can affect the pathway of the reaction as well. Polar protic solvents may be used to hinder nucleophiles, thus disfavoring E2 / SN2 from occurring.
Acid catalyzed dehydration of secondary / tertiary alcohols
We’ll take a look at a mechanism involving solvolysis during an E1 reaction of cyclohexanol in sulfuric Acid.
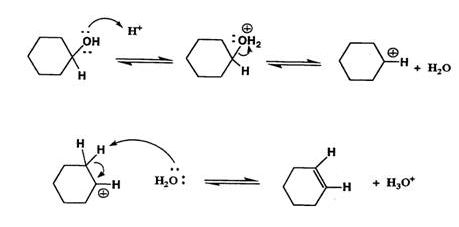
- Step 1: The OH group on the cyclohexanol is hydrated by H2SO4, represented as H+. This allows the OH to become an H2O, which is a better leaving group.
- Step 2: Once the OH has been protonated, the H2O molecule leaves via a heterolysis step, taking its electrons with it. This creates a carbocation intermediate on the attached carbon.
- Step 3: Another H2O molecule comes in to deprotonate the beta carbon, which then donates its electrons to the neighboring C-C bond. The carbons are rehybridized from sp3 to sp2, and thus a pi bond is formed between them.
Mechanism for Alkyl Halides
This mechanism is a common application of E1 reactions in the synthesis of an alkene.
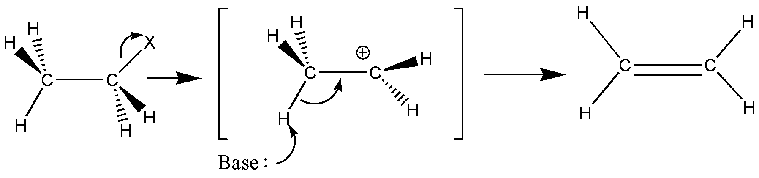
Once again, we see the basic 2 steps of the E1 mechanism.
- The leaving group leaves along with its electrons to form a carbocation intermediate.
- A base deprotonates a beta carbon to form a pi bond.
How are regiochemistry & stereochemistry involved?
In some cases we see a mixture of products rather than one discrete one. This can happen whenthe carbocation has two or more nearby carbons that are capable of being deprotonated. In many cases one major product will be formed, the most stable alkene. In terms of regiochemistry, Zaitsev’s rule states that when more than one product can be formed, the more substituted alkene is the major product.
Unlike E2 reactions, E1 is not stereospecific. Thus, a hydrogen is not required to be anti-periplanar to the leaving group.
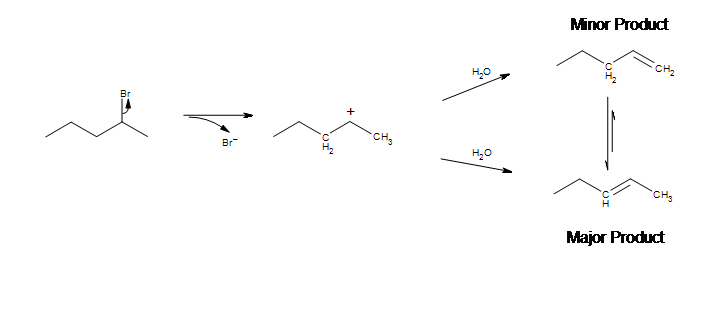
In this example, we can see two possible pathways for the reaction. One in which the methyl on the right is deprotonated, and another in which the CH2 on the left is deprotonated. Either pathway leads to a plausible product, but it turns out that pent-2-ene is the major product. As stated by Zaitsev’s rule, deprotonation will mainly happen at the most substituted carbon to form the more substituted (and more stable) alkene. In practice, the pent-2-ene product will be formed as a mixture of cis and trans alkenes, with the trans being the major isomer since it is more stable; only the trans is shown in the figure above.
Key features of the E1 elimination
The main features of the E1 elimination are:
- It usually uses a weak base (often ROH) with an alkyl halide, or it uses an alcohol in the presence of H2SO4 or H3PO4.
- Only secondary or tertiary alkyl halides are effective reactants, with tertiary reacting most easily. Heat is often used to minimize competition from SN1.
- The reaction is not stereoselective, so cis/trans mixtures are usual.
- Zaitsev’s Rule applies, so the more substituted alkene is usually major.
Outside links
Khan Academy video on E1
Problems
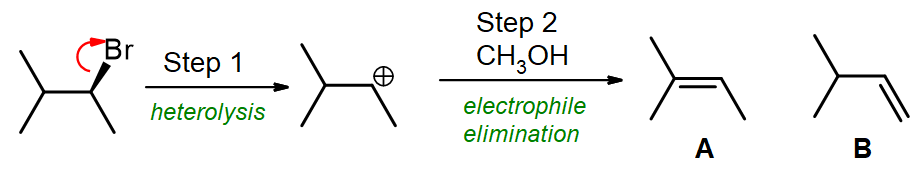
1) 3-Bromo-2-methylbutane is heated with methanol and an E1 elimination is observed.
(a) Which of these steps is the rate determining step (step 1 or step 2)?
(b) Which alkene is the major product formed (A or B)?
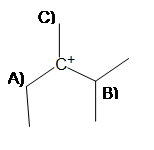
2) In order to produce the most stable alkene product, from which carbon should the base deprotonate (A, B, or C)?
If the carbocation were to rearrange, on which carbon would the positive charge go onto without sacrificing stability (A, B, or C)?
3) Predict the major product of the following reaction.
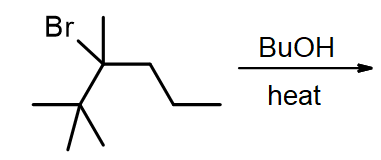
4) (True or False) – There is no way of controlling the product ratio of E1 / Sn1 reactions.
5) Explain why the presence of a weak base / nucleophile favors E1 reactions over E2.
Answers
[reveal-answer q=”231323″]Show Answer[/reveal-answer]
[hidden-answer a=”231323″]
1. Step 1, alkene A
2. B, B
3. 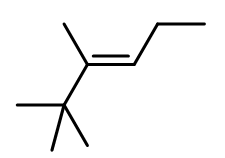
4. False – They can be thermodynamically controlled to favor a certain product over another.
5. By definition, an E1 reaction is a Unimolecular Elimination reaction. This means the only rate determining step is that of the dissociation of the leaving group to form a carbocation. Since E2 is bimolecular and the nucleophilic attack is part of the rate determining step, a weak base/nucleophile disfavors it and ultimately allows E1 to dominate. (Don’t forget about SN1 which still pertains to this reaction simaltaneously).
[/hidden-answer]
Videos
Outside Sources
- Vollhardt, K. Peter C., and Neil E. Schore. Organic Chemistry Structure and Function. New York: W. H. Freeman, 2007.
- McMurry, J., Simanek, E. Fundamentals of Organic Chemistry, 6th edition. Cengage Learning, 2007.
- 14.3: Elimination by the E1 and E2 mechanisms. Authored by: Tim Soderberg (University of Minnesota, Morris). Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Book%3A_Organic_Chemistry_with_a_Biological_Emphasis_(Soderberg)/14%3A_Reactions_with_stabilized_carbanion_intermediates%2C_part_II/14.3%3A_Elimination_by_the_E1_and_E2_mechanisms. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- E2 Reactions. Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Supplemental_Modules_(Organic_Chemistry)/Reactions/Elimination_Reactions/E2_Reactions. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- E1 Reactions. Authored by: Satish Balasubramanian. Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Supplemental_Modules_(Organic_Chemistry)/Reactions/Elimination_Reactions/E1_Reactions. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 12.3: Comparing E1 and E2 Mechanisms. Authored by: William Reusch. Located at: https://chem.libretexts.org/LibreTexts/Purdue/Purdue%3A_Chem_26605%3A_Organic_Chemistry_II_(Lipton)/Chapter_12._Elimination_Reactions/12.3%3A_Comparing_E1_and_E2_Mechanisms. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike