
8.3: Factors affecting rate of nucleophilic substitution reactions
- Page ID
- 225804
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Designing a “good” nucleophilic substitution
If you want to do well in this class, there are several things you need to work hard at: Being attentive in class, studying the notes and this textbook (especially before exams), practicing problems, and completing the quizzes and homeworks. As long as you do all of these things then you’re likely to pass (though I can’t give guarantees!). So there are many different factors that can affect your grade. In the same way, the outcome of a reaction (such as nucleophilic substition) depends on many different things – reactants, solvent, etc. When we want to make a chemical in a lab or on a chemical plant, we need to design the reaction so that it works well, and gives a good yield of the product in a reasonable time. The reactants and conditions we use will depend on what we’re trying to do. In this section, we examine what factors will help an SN2 or SN1 reaction be successful.
Factors affecting the SN2 reaction
As we saw in the previous section, in the SN2 reaction the rate of reaction depends on both the electrophile (usually an alkyl halide) and the nucleophile.
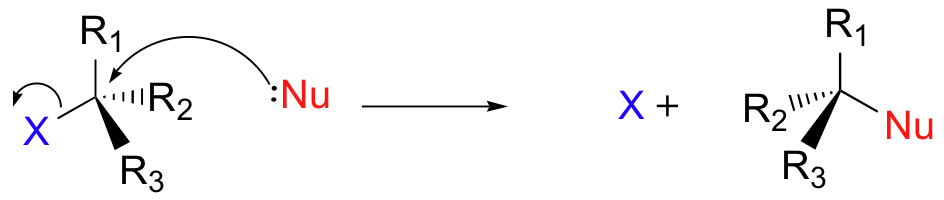
In practice, the rates of SN2 reactions vary enormously, and for a practicable procedure we need to make sure that the reaction will happen at a reasonable rate. So what makes for a good SN2 reaction? We need to consider what makes a suitable nucleophile, and what makes a suitable electrophile.
Nucleophile strength
In section 6.5, we learnt what makes a nucleophile strong (reactive) or weak (unreactive). Anything which removes electron-density from the nucleophilic atom will make it less nucleophilic. We summarized the main points from 6.5 as follows:
- Charge – negatively charged => stronger nucleophile
- Within a row – more electronegative atom => weaker nucleophile
- Within a column, size of atom. Polar protic solvent, bigger atom is better; polar aprotic solvent, smaller atom is better.
- Resonance – if the nucleophilic lone pair can be delocalized by resonance, it will make it less nucleophilic
- Steric hindrance – a hindered nucleophilic atom will tend to be less reactive, particularly when attacking a crowded electrophile.
Regarding the solvent, polar aprotic solvents such as DMSO, DMF, acetone or acetonitrile are popular choices for SN2 reactions, because rates are generally faster than with polar protic solvents (water, alcohols, etc.). This is because the nucleophile is almost “naked” in aprotic solvents, whereas in polar protic solvents it is surrounded by a cage of solvent molecules.
If we have a strong nucleophile, the SN2 reaction will happen faster; a weak nucleophile will react more slowly and may not even react. So in general we want a strong nucleophile.
The electrophile
(a) Structure of the alkyl group
In the structure of the SN2 transition state, there are 90o bond angles between the breaking bond to the leaving group and the three bonds which remain connected to the carbon as well as between the bond being made to the nucleophile and those same three bonds.
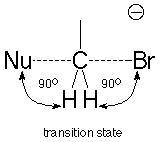
As long as the two of the groups attached to the carbon being attacked are small hydrogens, the repulsions which happen do not require much energy. If the groups attached to the carbon are larger, though, like methyl groups, the transition state energy increases, the activation energy increases, and the reaction becomes much slower.
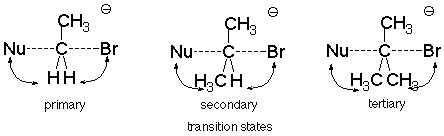
This means that the reactivity order for alkyl halides in SN2 reactions is:
methyl > primary > secondary > tertiary
The practical outcome of this is that SN2 reactions are generally reliable only when the alkyl halide is primary, though under the correct conditions secondary halides can react well also.
(b) Leaving group ability – what makes a good leaving group?
In our general discussion of nucleophilic substitution reactions, we have until now been designating the leaving group simply as “X”. As you may imagine, however, the nature of the leaving group is an important consideration: if the C-X bond does not break, the new bond between the nucleophile and electrophilic carbon cannot form, regardless of whether the substitution is SN1 or SN2. There are two main factors: The strength of the C-X bond, and the stability of the X group after it has left. It turns out that the two factors lead to the same prediction for halogen leaving group ability:
I > Br > Cl > F
C-X bond strength
Since the bond between the carbon and the leaving group is being broken in the transition state, the weaker this bond is the lower the activation energy and the faster the reaction. This leads to the following reactivity order for alkyl halides
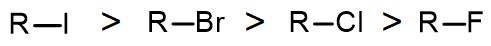 RBr>RCl>RF” width=”321″ height=”30″>
RBr>RCl>RF” width=”321″ height=”30″>
Practically, alkyl fluorides are not used for SN2 reactions because the C-F bond is too strong. Often alkyl iodides are reactive enough to be difficult to store, so the the common choices for reactions are alkyl chlorides and alkyl bromides.
Stability of the group after leaving
When the C-X bond breaks in a nucleophilic substitution, the pair of electrons in the bond goes with the leaving group. In this way, the leaving group is analogous to the conjugate base in a Brønsted-Lowry acid-base reaction. When we were evaluating the strength of acids in chapter 7, what we were really doing was evaluating the stability of the conjugate base that resulted from the proton transfer. All of the concepts that we used to evaluate the stability of conjugate bases we can use again to evaluate leaving groups. In other words, the trends in basicity are parallel to the trends in leaving group potential – the weaker the base, the better the leaving group. Just as with conjugate bases, the most important question regarding leaving groups is this: when a leaving group leaves and takes a pair of electrons with it, how well is the extra electron density stabilized?
In laboratory synthesis reactions, halides often act as leaving groups. Iodide, which is the least basic of the four main halides, is also the best leaving group – it is the most stable as a negative ion. Fluoride is the least effective leaving group among the halides, because fluoride anion is the most basic.
We already know that the use of polar, aprotic solvents increases the reactivity of nucleophiles in SN2 reactions, because these solvents do not ‘cage’ the nucleophile and keep it from attacking the electrophile.
Factors favoring SN2
To design an effective SN2 reaction using an alkyl halide, we need:
- An unhindered alkyl halide (preferably methyl or primary, but secondary may be possible)
- A good leaving group (preferably I or Br)
- A strong nucleophile
- A suitable solvent – polar aprotic is most effective
Factors affecting the SN1 reaction
As we learnt in section 8.2, the nucleophile has no effect on the rate of an SN1 reaction. This means that we only need to consider the electrophile, usually an alkyl halide. Another feature of the SN1 reaction is that it is often prone to side reactions, which is why it is less used in synthesis than the SN2 reaction.
The electrophile
This topic was examined in general in section 6.5., and also considered above for SN2. But a electrophile that is good for SN2 is not necessarily good for SN1, for reasons that will become clear. We also have a new factor to consider – the stability of the carbocation that is formed as a result of the heterolysis step.
(a) Structure of the alkyl group
In the vast majority of the nucleophilic substitution reactions you will see in this and other organic chemistry texts, the electrophilic atom is a carbon which is bonded to an electronegative atom, usually oxygen, nitrogen, sulfur, or a halogen. The concept of electrophilicity is relatively simple: an electron-poor atom is an attractive target for something that is electron-rich, i.e. a nucleophile. However, we must also consider the effect of steric hindrance on electrophilicity. In addition, we must discuss how the nature of the electrophilic carbon, and more specifically the stability of a potential carbocationic intermediate, influences the SN1 reaction.
Steric effects on electrophilicity
In an SN1 mechanism, the nucleophile attacks an sp2-hybridized carbocation intermediate, which has trigonal planar geometry with ‘open’ 120 angles.

With this open geometry, the empty p orbital of the electrophilic carbocation is no longer significantly shielded from the approaching nucleophile by the bulky alkyl groups. A carbocation is a very potent electrophile, and the nucleophilic step occurs very rapidly compared to the first (ionization) step. This is in direct contrast to the SN2 reaction, where bulky alkyl groups hinder the reaction.
Stability of carbocation intermediates
We know that the rate-limiting step of an SN1 reaction is the first step – formation of the this carbocation intermediate. The rate of this step – and therefore, the rate of the overall substitution reaction – depends on the activation energy for the process in which the bond between the carbon and the leaving group breaks and a carbocation forms. According to Hammond’s postulate (section 5.5), the more stable the carbocation intermediate is, the faster this first bond-breaking step will occur. In other words, the likelihood of a nucleophilic substitution reaction proceeding by a dissociative (SN1) mechanism depends to a large degree on the stability of the carbocation intermediate that forms.
We previously considered carbocation stability in section 6.5., and we found that in general, more substituted carbocations are more stable:
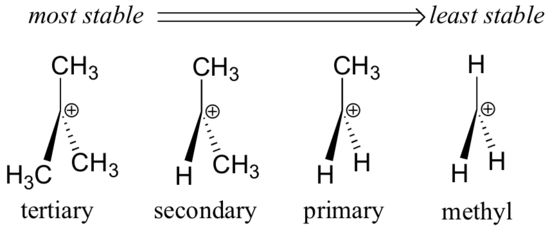
A positively charged species such as a carbocation is very electron-poor, and thus anything which donates electron density to the center of electron poverty will help to stabilize it. Conversely, a carbocation will be destabilized by an electron withdrawing group.
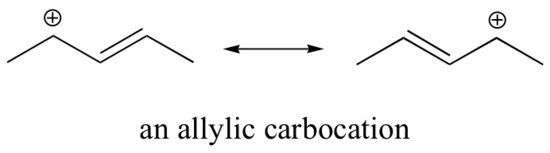
Stabilization of a carbocation can also occur through resonance, which allows the “burden” of the negative charge to be delocalized, or shared, onto more than one atom. Resonance effects as a rule are more powerful than inductive effects. For example, a cation next to a double bond will delocalize the charge via type IV resonance, so an allylic carbocation is more stable than a comparable one that cannot do resonance:
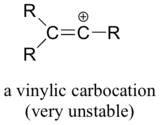
Finally, vinylic carbocations, in which the positive charge resides on a double-bonded carbon, are very unstable and thus unlikely to form as intermediates in any SN1 reaction.
When considering the possibility that a nucleophilic substitution reaction proceeds via an SN1 pathway, it is critical to evaluate the stability of the hypothetical carbocation intermediate. If this intermediate is not sufficiently stable, an SN1 mechanism must be considered unlikely, and the reaction probably proceeds by an SN2 mechanism.
Leaving group
The C-X bond breaks in the rate determining step of SN1, just as it does in SN2, and in fact the rules are the same for determining a “good” leaving group. Again these are determined by the C-X bond strength and the stability of X after it has left. This means that we see the same trends as we did for SN2, where the larger halogens make better leaving groups, i.e.,
I > Br > Cl > F
Side reactions in SN1
(a) Elimination
In all of our discussion so far about nucleophilic substitutions, we have ignored another important possibility. In many cases, including the two examples above, substitution reactions compete with a type of reaction known as elimination. This will be covered in detail soon, in section 8.5. Consider, for example, the two courses that a reaction could take when 2-bromo-2-methylpropane reacts with water:

We begin with formation of the carbocation intermediate. In pathway ‘a’, water acts as a nucleophile – this is, of course, the familiar SN1 reaction. However, a water molecule encountering the carbocation intermediate could alternatively act as a base rather than as a nucleophile, plucking a proton from one of the methyl carbons and causing the formation of a new carbon-carbon p bond. This alternative pathway is called an elimination reaction, and in fact with the conditions above, both the substitution and the elimination pathways will occur in competition with each other. Elimination can be minimized by keeping the reaction cold, but some of this side-reaction is often inevitable.
(b) Carbocation rearrangements
These will be covered very soon, in section 8.4. If the carbocation can easily rearrange to a more stable carbocation, then rearrangement products are likely to be important, and the reaction may lead to mixtures.
Solvent
The rates of SN1 reactions are generally increased by the use of a highly polar solvent, including protic (hydrogen bonding) solvents such as water or ethanol. In essence, a protic solvent increases the reactivity of the leaving group in an SN1 reaction, by helping to stabilize the products of the first (ionization) step. In the SN1 mechanism, remember, the rate determining step does not involve the nucleophilic species, so any reduction of nucleophilicity does not matter. What matters is that the charged products of the first step – the carbocation intermediate and the anionic leaving group – are stabilized best by a highly polar, protic solvent.
Factors favoring SN1
To design an effective SN1 reaction using an alkyl halide, we need:
- A highly substituted alkyl halide (preferably tertiary or resonance-stabilized, but secondary may be possible), ideally one which will not lead to rearrangement
- A good leaving group (preferably I or Br)
- A non-basic nucleophile (to reduce the elimination side reaction)
- A suitable solvent – polar protic is most effective
Predicting SN1 vs. SN2 mechanisms
When considering whether a nucleophilic substitution is likely to occur via an SN1 or SN2 mechanism, we really need to consider three factors:
1) The electrophile: when the leaving group is attached to a methyl group or a primary carbon, an SN2 mechanism is favored (here the electrophile is unhindered by surrounded groups, and any carbocation intermediate would be high-energy and thus unlikely). When the leaving group is attached to a tertiary, allylic, or benzylic carbon, a carbocation intermediate will be relatively stable and thus an SN1 mechanism is favored.
2) The nucleophile: powerful nucleophiles, especially those with negative charges, favor the SN2 mechanism. Weaker nucleophiles such as water or alcohols favor the SN1 mechanism.
3) The solvent: Polar aprotic solvents favor the SN2 mechanism by enhancing the reactivity of the nucleophile. Polar protic solvents favor the SN1 mechanism by stabilizing the carbocation intermediate. SN1 reactions are frequently solvolysis reactions.
For example, the reaction below has a tertiary alkyl bromide as the electrophile, a weak nucleophile, and a polar protic solvent (we’ll assume that methanol is the solvent). Thus we’d confidently predict an SN1 reaction mechanism. Because substitution occurs at a chiral carbon, we can also predict that the reaction will proceed with racemization.

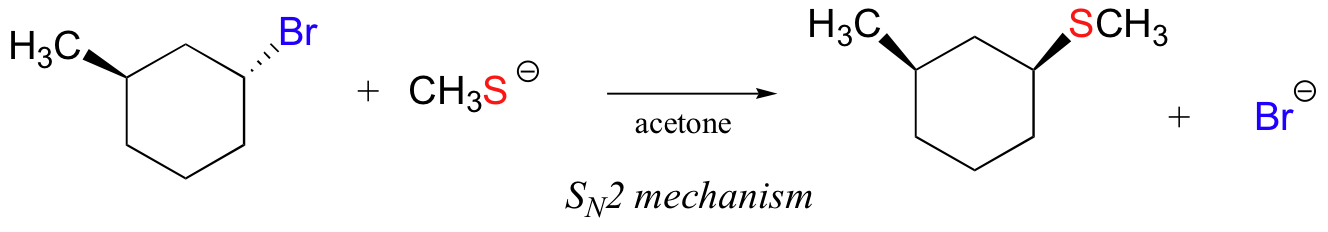
In the reaction below, on the other hand, the electrophile is a secondary alkyl bromide – with these, both SN1 and SN2 mechanisms are possible, depending on the nucleophile and the solvent. In this example, the nucleophile (a thiolate anion) is strong, and a polar protic solvent is used – so the SN2 mechanism is heavily favored. The reaction is expected to proceed with inversion of configuration.
Exercise
Determine whether each substitution reaction shown below is likely to proceed by an SN1 or SN2 mechanism.
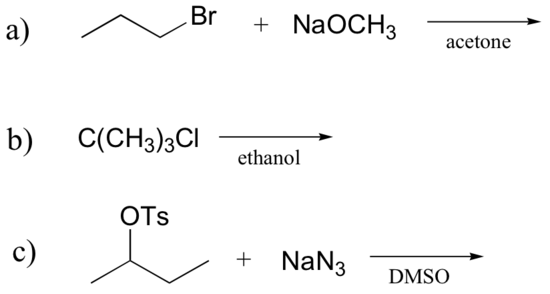
[reveal-answer q=”778981″]Show Solution[/reveal-answer]
[hidden-answer a=”778981″]
a) SN2 (primary electrophile, strong nucleophile, polar aprotic solvent)
b) SN1 (tertiary electrophile, weak nucleophile, protic solvent)
c) SN2 (secondary electrophile, strong nucleophile, polar protic solvent)
[/hidden-answer]
- 24: Nucleophilic Substitution, SN2, SN1. Authored by: Kirk McMichaelu00a0(Washington State University). Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Book%3A_Organic_Chemistry_-_A_%22Carbonyl_Early%22_Approach_(McMichael)/24%3A_Nucleophilic_Substitution%2C_SN2%2C_SN1. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 8.3: More about nucleophiles. Authored by: Tim Soderberg, (University of Minnesota, Morris). Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Book%3A_Organic_Chemistry_with_a_Biological_Emphasis_(Soderberg)/Chapter_08%3A_Nucleophilic_substitution_reactions_I/8.3%3A_More_about_nucleophiles. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 8.5: Leaving groups. Authored by: Tim Soderberg (University of Minnesota, Morris). Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Book%3A_Organic_Chemistry_with_a_Biological_Emphasis_(Soderberg)/Chapter_08%3A_Nucleophilic_substitution_reactions_I/8.5%3A_Leaving_groups. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 8.4: Electrophiles and carbocation stability. Authored by: Tim Soderberg (University of Minnesota, Morris). Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Book%3A_Organic_Chemistry_with_a_Biological_Emphasis_(Soderberg)/Chapter_08%3A_Nucleophilic_substitution_reactions_I/8.4%3A_Electrophiles_and_carbocation_stability. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- 25: Elimination - E2 and E1. Authored by: Kirk McMichael (Washington State University). Located at: https://chem.libretexts.org/Textbook_Maps/Organic_Chemistry/Book%3A_Organic_Chemistry_-_A_%22Carbonyl_Early%22_Approach_(McMichael)/25%3A_Elimination_-_E2_and_E1. Project: Chemistry LibreTexts. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike
- Organic Chemistry with a Biological Emphasis Volume I. Authored by: Timothy Soderberg. Provided by: University of Minnesota, Morris. Located at: https://digitalcommons.morris.umn.edu/chem_facpubs/1. License: CC BY-NC-SA: Attribution-NonCommercial-ShareAlike