
1.25: Elimination - E2 and E1
- Page ID
- 81791

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)SN1 Energy and Kinetics
Last time we saw an overview of the nucleophilic substitution mechanisms of alkyl halides. We examined one of these, the SN2 mechanism in detail. Today we'll examine the other, the SN1 mechanism, and then go on to look at elimination reactions, the major competition for substitutions. Here's the outline of the SN1 mechanism:
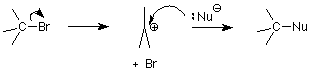
Recalling what the "2" in SN2 meant -- that the reaction was second order, two molecules had to collide to provide the activation energy needed to reach and pass through the transition state -- we can guess that the "1" in SN1 means that only one molecule needs to be "activated" in order to reach the transition state. That molecule is the alkyl halide. The critical step in this mechanism is the first step, in which the bond between the carbon atom and the halogen leaving group is broken. The transition state for this step has the bond stretched far enough that the halide ion is balanced between leaving as a stable chloride or bromide ion or slipping back into a covalent bond.
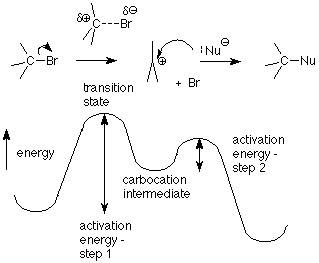
(The energy required to break this bond comes from random collisions with the solvent without the solvent reacting.) The activation energy for the first step is higher than for the second step, so the rate of the reaction is controlled by how many molecules get through the first step. Once a molecule is through the first step, it can react rapidly in the second step. We call the step which is slowest the "rate determining step." Notice that the rate determining step for this reaction doesn't involve the nucleophile. That means that changing the concentration of the nucleophile doesn't affect the rate. The only concentration which affects the rate is the concentration of the alkyl halide. We thus have a first order reaction:
Rate = k[RX]
This also means that the strength of the nucleophile -- its ability to use its electron pair to make a bond -- isn't important in determining how fast the reaction goes. It's not involved in the rate determining step, so it has no effect on the energy of that transition state.
Effect of Alkyl Group Structure
What does affect the energy of the rate determining transition state? If we examine its structure in more detail, we notice that there is considerable positive charge on the carbon atom and the carbon-halogen bond is nearly broken.
The nearly broken bond tells us that the effect of changing the halogen is the same as it was for the SN2 reaction:
RI > RBr > RCl > RF
The substantial degree of positive charge on the carbon is important in explaining how the structure of the alkyl group affects the rate. A large number of experiments have established that the reactivity order for alkyl halides in the SN1 mechanism is:
tertiary > secondary > primary > methyl
This is just the opposite of the order for the SN2 reaction. The outcome of this contrast is that tertiary alkyl halides consistently use the SN1 pathway. Primary and methyl alkyl halides use th SN2 pathway. For secondary alkyl halides either pathway is possible, and we have to look at other information to make a decision.
The reactivity order tells us that the transition state for the SN1 reaction of a tertiary alkyl halide has a lower energy than the transition state for a correspinding secondary alkyl halide. Perhaps the easiest way to understand this is to recognize that the transition state, with its substantial positive charge on carbon, is very much like the carbocation intermediate which is formed in the first step. Changes which make the carbocation intermediate, with its positively charged carbon, more stable will also make the transition state, with its nearly positively charged carbon, more stable.
The major thing which determines the energy of the carbocation intermediate is that it has only six electrons in its valence shell. It is an electrophile, a Lewis acid, and a seriously electron deficient molecule. The more electron density which can be shifted towards the positively charged carbon, the lower the energy. In a tertiary carbocation, there are three carbon atoms, each bonded to three other atoms, connected to the electron deficient carbocation carbon. This adds up to 18 electrons in the bonds adjacent to the carbocation, all of which can shift slightly to help neutralize the positive charge. Contrast this to the situation in a methy carbocation. Here there are no valence electrons other than the ones holding the hydrogens to the carbon, so there is a very poor supply of electrons to assist with lowering the energy of the carbocation.
While this discussion has focused on the carbocation intermediate with its full positive charge, the same principles apply in the case of the partial positive charge in the transition state. The more electrons there are in the near neighborhood, the more stable the transition state. There are more electrons available on the carbon atoms attached to a tertiarly carbocation center than there are on the hydrogens attached to a methyl carbocation center.
If there are pi bonds involved with a carbon attached to the carbocation carbon, the energy is reduced even more. Recall that pi electrons are less tightly bound than sigma electrons, so they are easier to move towards the electron deficiency. This can be symbolized in resonance terms:
The effect of this is that alkyl halides which have carbon-carbon pi bonds located one atom away from the carbon bearing the halogen are quite reactive in SN1 reactions.
Stereochemical Outcome
The product of an SN1 reaction is formed in the second step. This step starts with the carbocation intermediate, so let's look at its structure to see what we can learn from it. First, notice the distinction between an intermediate and a transition state. Remember that a transition state is at a maximum energy. Any slight change will cause it to fall from it's precarious perch and become either the product or the reactant of its step. In contrast, an intermediate is at an energy minimum so that most small changes only push it a little way up a hill from its lowest energy situation. It takes a substantial collision to provide it with the energy needed to reach a transition state so as to pass through and become a product. If the activation energy is fairly small, as it is with the second step of the SN1 mechanism, the reaction may well be fast, but it doesn't occur with every small wiggle as it does with a transition state.
Another way to say this is that a transition state has a very, very short lifetime, while an intermediate may exist for micro- or milliseconds, which is a long time and provides opportunities for many collisions with other molecules.
One outcome of this is that the carbocation intermediate "lives" long enough for a nucleophile to approach it on either face of the molecule. The carbocation is flat with 120o angles between its three bonds. That means that it is trigonal like the carbon of a carbonyl group and is sp2 hybridized. The "empty" orbital which we can associate with the electrophilic characteristics of the carbocation is a p orbital.
If the carbocation intermediate has the opportunity to engage in many collisions with potential nucleophiles, there is an equal chance that a nucleophile will attack at one lobe or the other of the p orbital. If there are three different groups attached to the carbocation center, the nucleophile will provide a fourth and generate a stereogenic carbon. Equal quantities of the two enantiomers will be expected so that the product mixture will not be optically active.
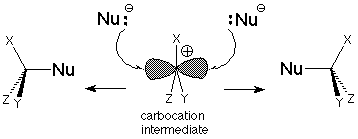
This is a somewhat idealized situation, since in practice the halide leaving group is "loitering" near one lobe of the p orbital. This makes reaction with the nucleophile easier on the other lobe, so there is usually some net inversion. The stereochemical oucome of the SN1 is not as clearcut as that of the SN2 mechanism's inversion, but it is still a distinguishing feature of the mechanism, one which can be used to decide which mechanism is operating in a particular reaction.
Solvolysis
The fact that the nucleophile is not involved in the rate determining step of an SN1 reaction also means that it proceeds well with a relatively weak nucleophiles. When the nucleophile is also the solvent, the reaction is called "solvolysis." Solvents like water and alcohols are particularly useful here, because they provide both a nucleophilic pair of electrons on the oxygen atom of the OH group and a fairly polar solvent which helps to stabilize the strongly polar transition state. The latter effect is much like the way in which a polar solvent dissolves a very polar substance like salt by surrounding its charged ions with polar solvent molecules.
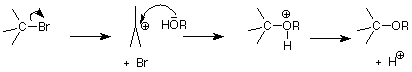
This process resembles the way in which an alcohol or water might attack the electrophilic carbon of a carbonyl group after an acid had added an H+ to the oxygen. Remember that in these reactions the neutral nucleophilic atom attacks first; then the H+ is lost.
A summary of the important differences between the SN1 and SN2 mechanisms is in Table 7.5 (Brown, p 189).
Competition with Elimination
One of the things we dealt with last time was the fact that nucleophiles are also Lewis bases. One outcome of this is that the same atom or group can attack a carbon in an SN1 or SN2 reaction -- behaving as a nucleophile -- or attack a hydrogen atom -- behaving as a Lewis base. The latter attack can lead to an elimination reaction. We will look at elimination reactions in more detail in a week or so, but we can usefully examine them as competitors for SN1 and SN2 reactions just now.
When we first learned about the Williamson ether synthesis, we learned that it works best when the alkyl halide is primary. We now understand that as a characteristic of the SN2 mechanism -- primary alkyl halides react faster than secondary or tertiary alkyl halides. However, a patient person might suggest that even a slow SN2 reaction might succeed if we were willing to wait a while. What defeats this strategy is that the alkoxide ion can also react as a base. This elimination reaction (an E2 reaction for future reference) is fast enough that it uses up the secondary or tertiary alkyl halide long before the much slower SN2 reaction produces any useful amount of product.
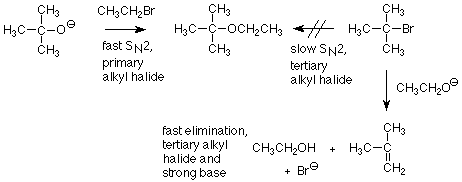
Elimination reactions are always potential competitors for substitution reactions. The key factors which alert us to situations favorable to eliminations are:
- An alkyl halide which is slow in SN2 reactions, i.e., tertiary and secondary alkyl halides.
- The presence of a strong base like an alkoxide or hydroxide ion.
These conditions will typically produce much more elimination product (an alkene) than substitution product.
Can we use an SN1 pathway to avoid this difficulty? Yes -- and the key here is that since the rate of an SN1 reaction is not sensitive to the concentration or strenght of the nucleophile we can avoid the strong bases which promote elimination. Solvolysis of a tertiary alkyl halide using an alcohol as both nucleophile and solvent can make an ether very effectively.
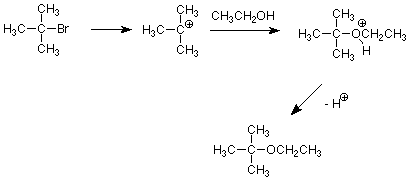
Of course, this is only possible when the desired nucleophile can be used as a solvent as is the case for alcohols and water.
SN1 and SN2 Reactions of Alcohols
To finish up today, let's revisit some reactions of alcohols and see if we can use the SN1 or SN2 pattern to understand them a little better. Recall that a useful method for making an alcohol into an alkyl halide was to treat the alcohol with the hydrogen halide, particularly when the alcohol was tertiary:
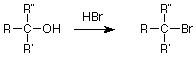
This looks like a nucleophilic substitution and since the alkyl group is tertiary, the SN1 pathway through a carbocation intermediate looks like a good guess. We can also identify the nucleophile as the bromide ion (Br-), but what about the leaving group? The obvious answer is that the OH- serves as the leaving group, but this is worrisome since we would be making a strong base in the presence of an acid. The solution appears when we remember that the unshared electron pairs on the alcohol oxygen are also weak bases. They can accept a proton (H+) from the strong acid HBr. When this is done, the leaving group is water, a weak base.
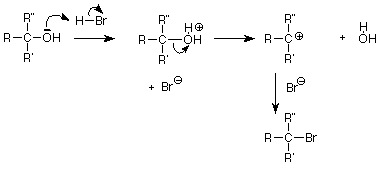
When the OH group of an alcohol is replaced by another nucleophile, we can be sure that the OH group is first converted to a good leaving group before the C-O bond is broken. That is the function of the H+ in an acid, the SO2 part of thionyl chloride and the phosphorus in PBr3. We won't concern ourselves with the details of these processes, but we can notice that the need to make the OH group into a good leaving group is the same whether the reaction is SN1 as we would expect for a tertiary alcohol or SN2 as we would expect for a primary alcohol.