
10.2: Pyridoxal Phosphate (Vitamin B6)
- Page ID
- 291189

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The coenzyme pyridoxal phosphate (commonly abbreviated PLP) is the active form of vitamin \(B_6\), or pyridoxine.
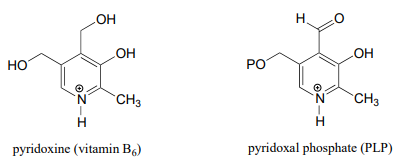
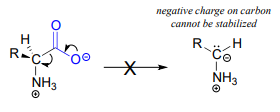
PLP is required for over 100 different reactions in human metabolism, primarily in the various amino acid biosynthetic and degradation pathways. The essential function of PLP is to act as an 'electron sink', stabilizing a negative formal charge that develops on key reaction intermediates. Some of reactions will be familiar to you from chapter 12 and 13 we will see examples, for instance, of -dependent \(\alpha \)-carbon racemization, as well as aldol- and Claisen-type reactions. Other reactions will be less familiar: for example, the participation of allows for decarboxylation of amino acids, a chemical step which would be highly unlikely without the coenzyme, and PLP is also required for a very important class of biochemical transformation called 'transamination', in which the amino group of an amino acid is transferred to an acceptor molecule. Before we dive into the reactions themselves, though, we need to begin by looking at a key preliminary step that is common to all of the PLP reactions we will see in this section.
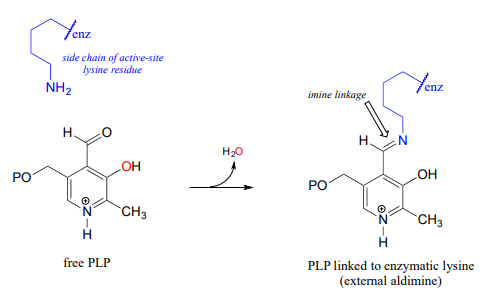
PLP in the active site: the imine linkage
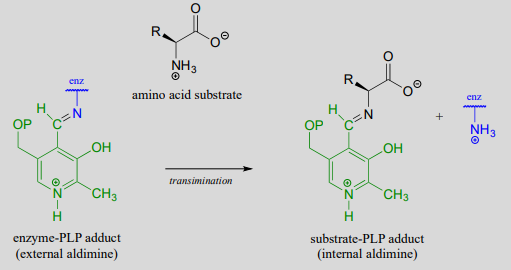
The common catalytic cycle of a PLP-dependent enzyme begins and ends with the coenzyme covalently linked to the enzyme's active site through an imine linkage between the aldehyde carbon of PLP and the amine group of a lysine residue (see section 10.5 to review the mechanism for imine formation). For a PLP-dependent enzyme to become active, a PLP molecule must first enter the active site of an enzyme and form an imine link to the lysine. This state is often referred to as an internal aldimine.
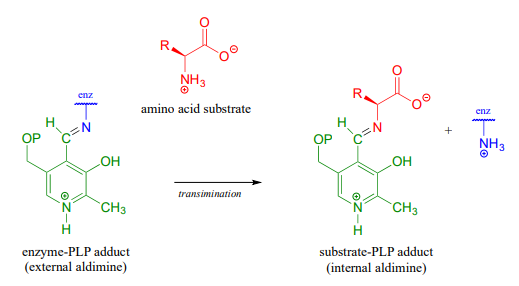
The first step of virtually all PLP-dependent reactions is transimination (section 10.5), as the amino group on the amino acid substrate displaces the amino group of the enzymatic lysine. This state - where the coenzyme is covalently linked to the substrate or product of the reaction - is often referred to as an external aldimine.
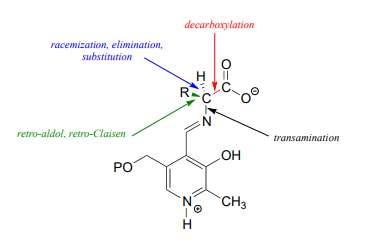
With the preliminary transimination accomplished, the real PLP chemistry is ready to start. The versatility of PLP in terms of its ability to assist with a wide variety of reaction types is illustrated by the figure below, showing how, depending upon the reaction/enzyme in question, PLP can assist in the cleavage of any one of the four bonds to the \(\alpha \)-carbon of the amino acid substrate.
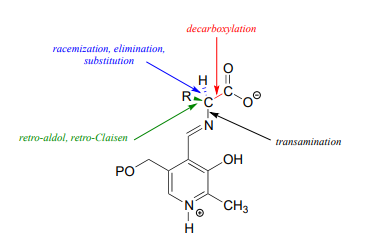
Let's look first at the reaction catalyzed by PLP-dependent alanine racemase. (EC 5.1.1.1).
PLP-dependent amino acid racemization
In section 12.2 we saw an example of a PLP-independent amino acid racemization reaction, in which the negatively-charged intermediate was simply the enolate form of a carboxylaten:
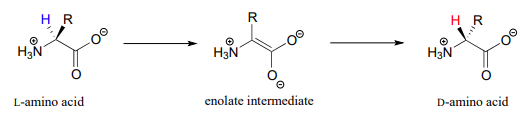
Many other amino acid racemase reactions, however, require the participation of PLP.
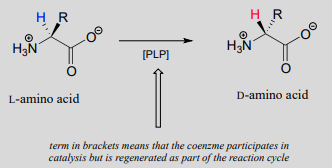
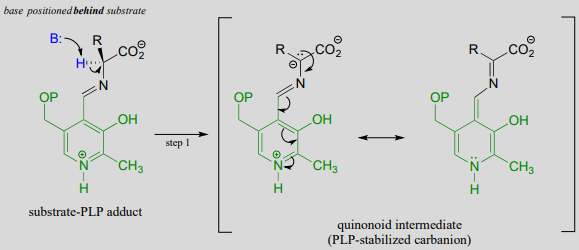
Like all other PLP-dependent reactions that we will see in this section, PLP-dependent amino acid racemization begins with a preliminary step in which the substrate becomes attached to the coenzyme through a transimination. Once it is linked to PLP in the active site, the a-proton of an amino acid substrate is abstracted by an active site base (step 1 below). The negative charge on the carbanion intermediate can, of course, be delocalized to the carboxylate group. The PLP coenzyme, however, provides an expanded network of conjugated \(\pi \)-bonds over which the electron density can be delocalized all the way down to the PLP nitrogen. This is what we mean when we say that the job of PLP is to act as an ‘electron sink’: the coenzyme is very efficient at absorbing, or delocalizing, the excess electron density on the deprotonated \(\alpha \)-carbon of the reaction intermediate. PLP is helping the enzyme to increase the acidity of the \(\alpha \)-hydrogen by stabilizing the conjugate base. A PLP-stabilized carbanion intermediate is commonly referred to as a quinonoid intermediate. Note that in the overall reaction equation below, PLP appears below the reaction arrow in brackets, indicating that it participates in the mechanism but is regenerated as part of the reaction cycle.
PLP-dependent amino acid racemization:
Mechanism:
Preliminary step - transimination
First step - deprotonation:
Second step - reprotonation from the opposite side:
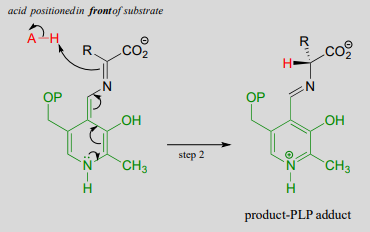
Final step - transimination:
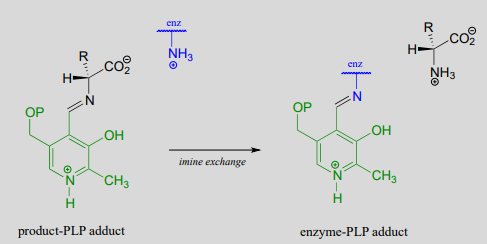
Just as in the PLP-independent racemase reactions, reprotonation occurs on the opposite side of the substrate (step 2), leading to the \(D\)-amino acid product.
All that remains is the final imine exchange which frees the \(D\)-amino acid product and re-attaches the coenzyme to the enzymatic lysine side-chain, ready to begin another catalytic cycle.
To simplify matters, from here on we will not include the preliminary and final transimination steps in our PLP reaction figures - we will only show mechanistic steps that occur while the substrate is attached to the coenzyme (the internal aldimine forms).
PLP-dependent decarboxylation
In the amino acid racemase reaction above, PLP assisted in breaking the \(\alpha \)-carbon to \(\alpha \)-proton bond of the amino acid. Other PLP-dependent enzymes can catalyze the breaking of the bond between the \(\alpha \)-carbon and the carboxylate carbon by stabilizing the resulting carbanion intermediate: these are simply decarboxylation reactions.
PLP-depended amino acid decarboxylation:
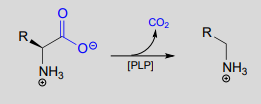
Mechanism:
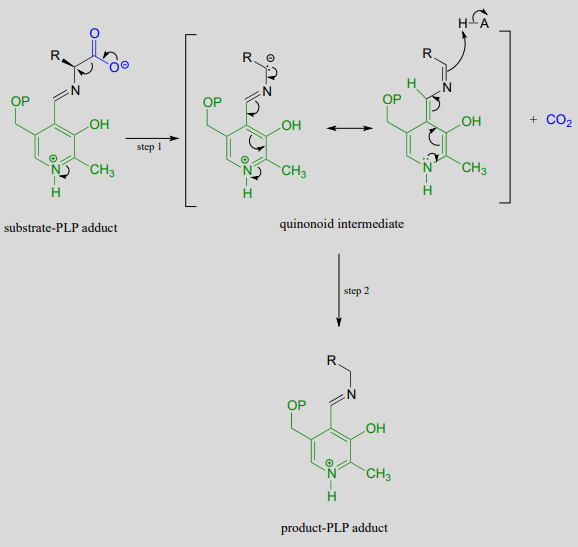
Notice something very important here: while in racemization reactions the assistance of PLP can be seen as 'optional' (in the sense that some racemase enzyme use PLP and others do not), the coenzyme is essential for amino acid decarboxylation steps. Without PLP, there is no way to stabilize the carbanion intermediate, and decarboxylation is not a chemically reasonable step.
One example of a PLP-facilitated decarboxylation reaction is the final step in the lysine biosynthesis pathway: (EC 4.1.1.20).
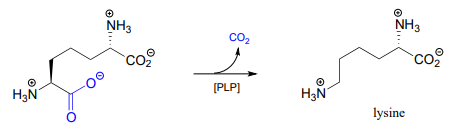
Draw mechanistic arrows for the carbon-carbon bond-breaking step of the PLP-dependent decarboxylation reaction above.
PLP-dependent retroaldol and retro-Claisen cleavage
(It would be a good idea before reading this section to review aldol/retro-aldol and Claisen/retro-Claisen reaction mechanisms in sections 12.3 and 13.3, respectively)
So far we have seen PLP playing a role in breaking the bond between the \(\alpha \)-carbon and its \(\alpha \)-proton (in the racemization reaction), and the bond between the \(\alpha \)-carbon and carboxylate carbon (in the decarboxylation reaction).
Other PLP-dependant enzymes catalyze cleavage of the bond between the \(\alpha\)-carbon and the first carbon on the amino acid side chain, otherwise known as the \(\beta \)-carbon. In the serine degradation pathway, serine is first converted to glycine by a retro-aldol cleavage reaction. (). Although a reasonable mechanism could be proposed without the participation of PLP, this reaction in fact requires the coenzyme to assist in stabilization of the negative charge on the carbanion intermediate.
A PLP-dependent retro-aldol cleavage reaction
(serine hydroxymethyl transferase, EC 2.1.2.1)
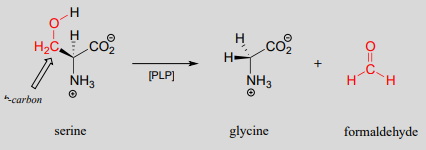
Mechanism:

Note that, in this reaction just as in the racemase reaction described previously, the key intermediate is a PLP-stabilized carbanion, or quinonoid.
What happens to the (toxic!) formaldehyde produced in this reaction? We will see later in this chapter how the serine hydroxymethyltransferase enzyme goes on to use another coenzyme called tetrahydrofolate to prevent the formaldehyde from leaving the active site and causing damage to the cell.
PLP also assists in retro-Claisen cleavage reactions (section 13.3), such as this step in the degradation of threonine. (EC 2.3.1.29)
A PLP-dependent retro-Claisen reaction:
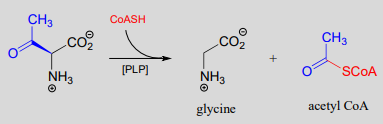
Mechanism:
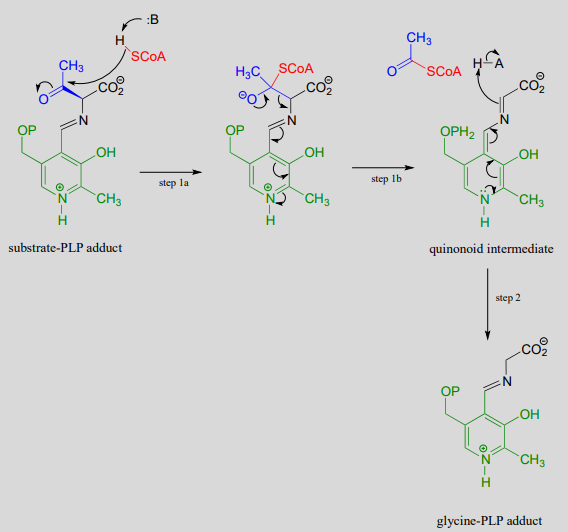
Notice how, like the retro-aldol reaction, the bond between the \(\alpha \)-carbon and the \(\beta \)-carbon of the amino acid substrate is broken (in step 1b).
PLP-dependent transamination
One of the most important reaction types in amino acid metabolism is transamination, in which an amino group on a donor molecule (often an amino acid) is transferred to a ketone or aldehyde acceptor molecule.
A transamination reaction:

Transamination phase 1 (transfer of amino group from amino acid substrate to coenzyme)
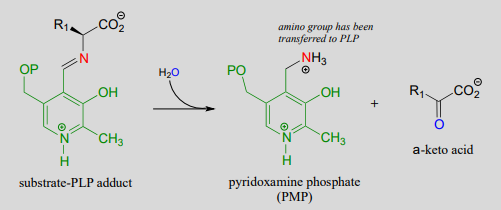
Mechanism:
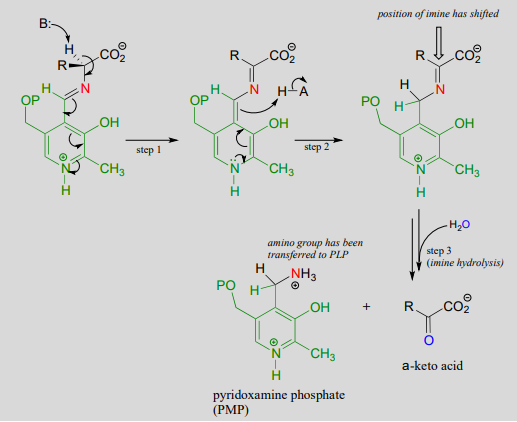
Once again, step 1 is abstraction of the \(\alpha \)-proton from the PLP-substrate adduct. However, in a transaminase reaction this initial deprotonation step is immediately followed by reprotonation at what was originally the aldehyde carbon of PLP (step 2 above), which results in a new carbon-nitrogen double bond (in other words, an imine) between the \(\alpha \)-carbon and the nitrogen atom of the original amino acid. The repositioned imine group is then hydrolyzed (step 3 above), breaking the carbon-nitrogen bond, transferring the amino group to the coenzyme, and releasing an \(\alpha \)-keto acid.
The coenzyme, which now carries an amine group and is called pyridoxamine phospate (\(PMP\)), next transfers the amine group to \(\alpha \)-ketoglutarate (to form glutamate) through a reversal of the whole process depicted above.
Transamination reaction, phase 2
(transfer of amino group from coenzyme to acceptor molecule)
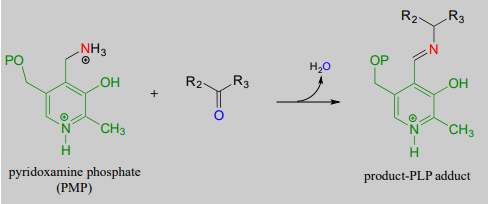
Mechanism:
see exercise below
In a transamination reaction, the PLP coenzyme not only provides an electron sink, it also serves as a temporary 'parking place' for an amino group as it is transferred from donor to acceptor.
Show a complete, step-by-step mechanism for 'phase 2' of the transamination reaction above.
Here is an example of a transamination reaction in the arginine biosynthesis pathway: EC 2.6.1.11
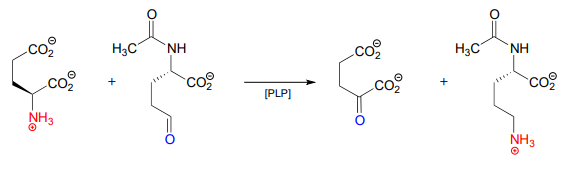
- Draw arrows for the first mechanistic step of 'phase 2' of the above transaminase reaction.
- Which carbon on the substrate side of the reaction will eventually become the \(\alpha \)-carbon of arginine?
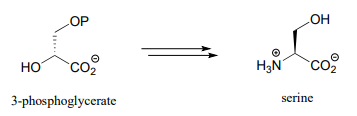
Propose a pathway, with three enzymatic steps, for the biosynthesis of serine from 3-phosphoglycerate. Include a generalized ('-ase') enzyme name for each step. Glutamate plays a role in the process as an amino group donor.
PLP-dependent \(beta \)-elimination and \(\beta \)-substitution
(Before starting this section, it would be a good idea to review \(E1cb\) \(\beta \)-elimination and conjugate addition reaction mechanisms in section 13.4)
By now it should be pretty apparent that PLP is a pretty versatile coenzyme! Two more reaction types in the PLP toolbox are \(\beta \)-elimination and \(\beta \)-substitution on amino acid substrates.
In a PLP-dependent \(\beta \)-elimination reaction, the coenzyme simply helps to stabilize the carbanion intermediate of the \(E1cb\) mechanism:
PLP-dependent \(\beta \)-elimination reaction
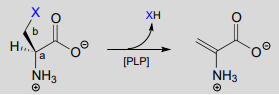
Mechanism:
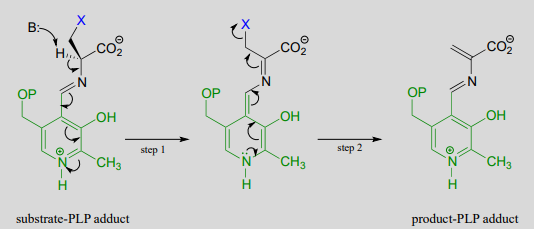
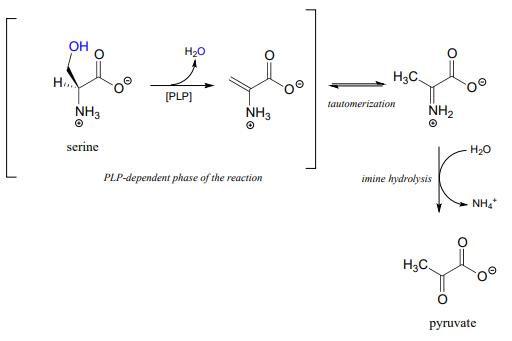
Serine dehydratase (EC 4.2.1.13) catalyzes a PLP-dependent \(\beta \)-elimination in the first step of the serine degradation pathway:
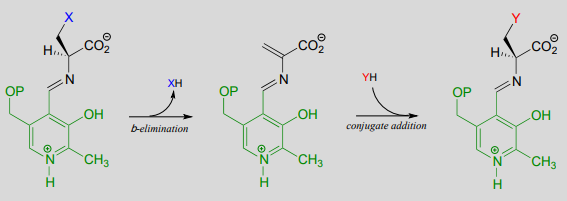
A \(\beta \)-substitution reaction is simply \(E1cb\) elimination followed directly by the reverse reaction (conjugate addition) with a different nucleophile (Y in the figure below):
A \(\beta \)-substitution reaction:
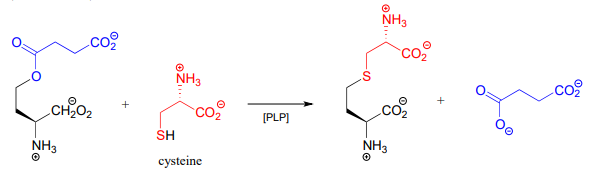
In many bacteria, the synthesis of cysteine from serine includes a PLP-dependent \(\beta \)-substitution step (EC 2.5.1.47).
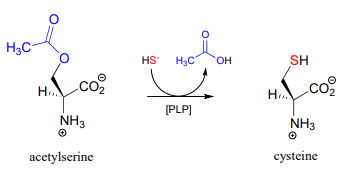
Draw a mechanism for the conjugate addition phase of the reaction above (end with the cysteine-PLP adduct).
PLP-dependent \(\gamma \)-elimination and \(\gamma \)-substitution reactions
The electron sink capability of PLP allows some enzymes to catalyze eliminations at the \(\gamma \)-carbon of some amino acid side chains, rather than at the \(\beta \)-carbon. secret to understanding the mechanism of a \(\gamma \)-elimination is that PLP essentially acts as an electron sink twice - it absorbs the excess electron density from not one but two proton abstractions.
\(\gamma \)-elimination
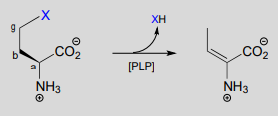
Mechanism:
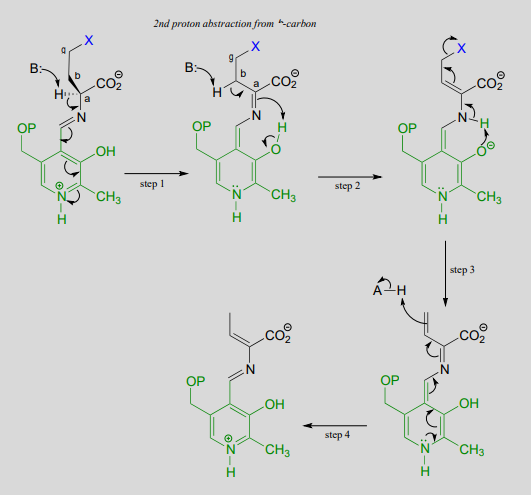
In a familiar first step, the \(\alpha \)-proton of the amino acid is abstracted by an enzymatic base, and the electron density is absorbed by PLP. Next comes the new part - before anything happens to the electron density from the first proton abstraction, a second proton, this time from the \(\beta \)-carbon on the side chain, is abstracted, forming an enamine intermediate (step 2). The phenolic proton on the pyridoxal ring of PLP donates a proton to the nitrogen. step 3, the leaving is expelled and a new \(\pi \)-bond forms between the \(\beta \) and \(\gamma \) carbons (step 3). This \(\pi \)-bond is short-lived, however, as the electron density from the first proton abstraction, which has been 'stored' in PLP all this time, flows back up to protonate the \(\alpha \)-carbon (step 4), leaving the \(\gamma \)-elimination product linked to PLP via the usual imine connection.
An example is the cystathionine \(\gamma \)-lyase reaction in the methionine degradation pathway (EC 4.4.1.1):
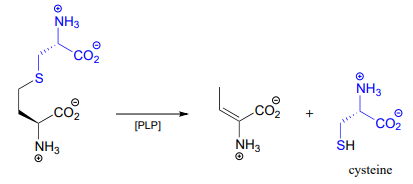
fig 2
A related reaction is PLP-dependent \(\gamma \)-substitution, which again is simply \(\gamma \)-elimination of a leaving group (X in the figure below) followed directly by the reverse process (a \(\gamma \)-addition) with a different nucleophile ('Nu' in the figure below).
PLP-dependent \(\gamma \)-substitution:
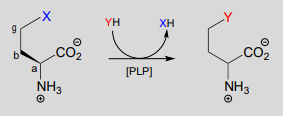
Mechanism:
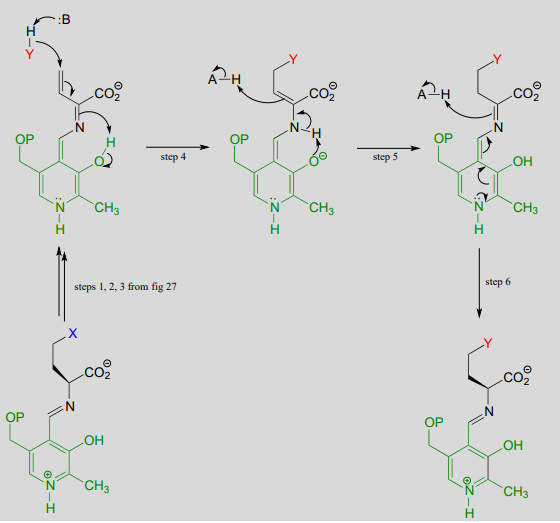
Below is a PLP-dependent \(\gamma \)-substitution reaction in the methionine degradation pathway (EC 4.2.1.22):
Racemase to aldolase: altering the course of a PLP reaction
We have seen how PLP-dependent enzymes catalyze a variety of reaction types - racemization, retroaldol/retro-Claisen cleavage, transaminination, elimination, and substitution - which, despite their apparent diversity, are all characterized by formation of a critical carbanion intermediate which is stabilized by the 'electron sink' property of the PLP coenzyme. Given this common mechanistic feature, it would be reasonable to propose that the active site architecture of these enzymes might also be quite close. This idea was nicely illustrated by an experiment in which researchers found that changing a single active site amino acid of PLP-dependent alanine racemase was sufficient to turn it into a retro-aldolase (J. Am. Chem. Soc. 2003, 125, 10158).
In the 'wild-type' (natural) alanine racemase reaction, an active site histidine (red in figure below) deprotonates a neighboring tyrosine residue (blue), which in turn acts as the catalytic base abstracting the \(\alpha \)-proton of the substrate. When researchers changed the tyrosine to an alanine (using a technique called 'site-directed mutagenesis'), and substituted \(\beta \)-hydroxytyrosine for the alanine substrate, the new 'mutant' enzyme catalyzed a retro-aldol reaction.
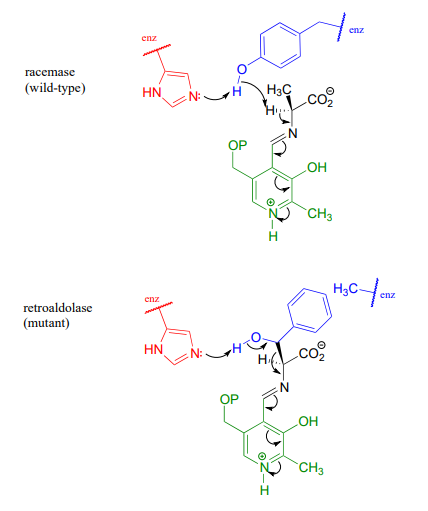
Notice what has happened here: the basic histidine, with no tyrosine to deprotonate because of the mutation, is instead positioned to abstract a proton from the \(\beta \)-hydroxyl group of the new substrate, setting up a retroaldol cleavage. That was all it took to change a racemase into a retroaldolase, because the necessary PLP electron sink system was all left in place. The researchers predicted correctly that the phenyl ring of \(\beta \)-hydroxy tyrosine would fit nicely in the space left empty due to the tyrosine to alanine change in the mutant enzyme's structure
These results underline the close mechanistic relationship between two PLP-dependent reactions which, at first glance, appear to be quite different - and suggest that PLP-dependent racemases and aldolases may have evolved from a common 'ancestor' enzyme.
Stereoelectronic considerations of PLP-dependent reactions
Recall that all PLP-dependent reactions involve the cleavage of one of the bonds coming from the \(\alpha \)-carbon of an amino acid substrate, with the coenzyme serving as an 'electron sink' to stabilize the intermediate that results. PLP-dependent enzymes accelerate this bond-breaking step by binding the substrate-PLP adduct in a conformation such that the bond being broken is close to perpendicular to the plane formed by the conjugated \(\pi \) system of PLP: this way, as the \(\alpha \)-carbon transitions from \(sp^3\) to \(sp^2\) hybridization, the unhybridized \(\pi \) orbital is already oriented to overlap with the rest of the conjugated system. For example, in alanine racemase the first step is cleavage of the \(C\alpha -H\) bond, so it must be that bond which is positioned near-perpendicular to the PLP plane:
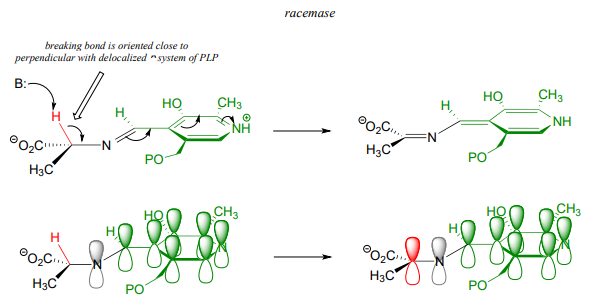
Likewise, in an amino acid decarboxylase, the \(C\alpha \)-carboxylate bond is held near-perpendicular to the PLP plane, and in hydroxymethyltransferase, the \(C\alpha -C\beta \) bond is in the perpendicular orientation:
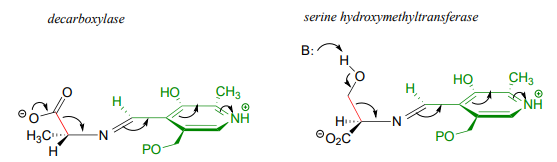
These are all good examples of how enzymatic catalysis is achieved, in part, by the ability of the active site to bind the substrate molecule in a specific conformation which contributes to the lowering of the activation energy of a key reaction step.