
4.3: Mass Spectrometry
- Page ID
- 234531

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Mass spectrometry (MS) is a powerful analytical technique widely used by chemists, biologists, medical researchers, and environmental and forensic scientists, among others. With MS, we are looking at the mass of a molecule, or of different fragments of that molecule.
The basics of a mass spectrometry experiment
There are many different types of MS instruments, but they all have the same three essential components. First, there is an ionization source, where the molecule is given a positive electrical charge, either by removing an electron or by adding a proton. Depending on the ionization method used, the ionized molecule may or may not break apart into a population of smaller fragments. In the figure below, some of the sample molecules remain whole, while others fragment into smaller pieces.
Next in line there is a mass analyzer, where the cationic fragments are separated according to their mass.
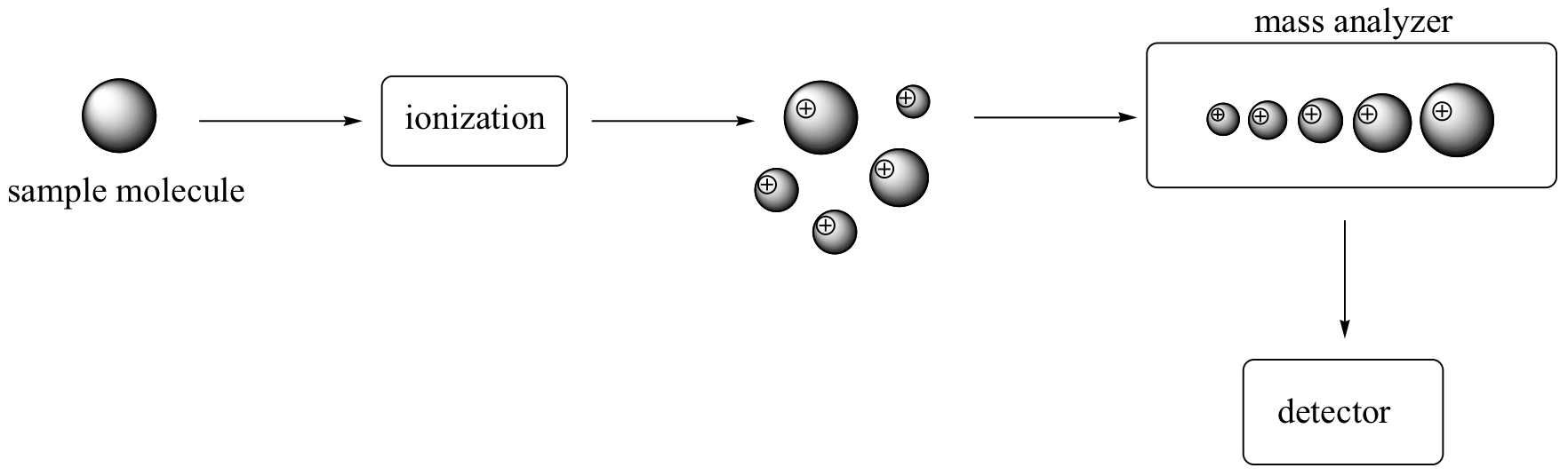
Finally, there is a detector, which detects and quantifies the separated ions.
One of the more common types of MS techniques used in the organic laboratory is electron ionization. In the ionization source, the sample molecule is bombarded by a high-energy electron beam, which has the effect of knocking a valence electron off of the molecule to form a radical cation. Because a great deal of energy is transferred by this bombardment process, the radical cation quickly begins to break up into smaller fragments, some of which are positively charged and some of which are neutral. The neutral fragments are either adsorbed onto the walls of the chamber or are removed by a vacuum source. In the mass analyzer component, the positively charged fragments and any remaining unfragmented molecular ions are accelerated down a tube by an electric field.
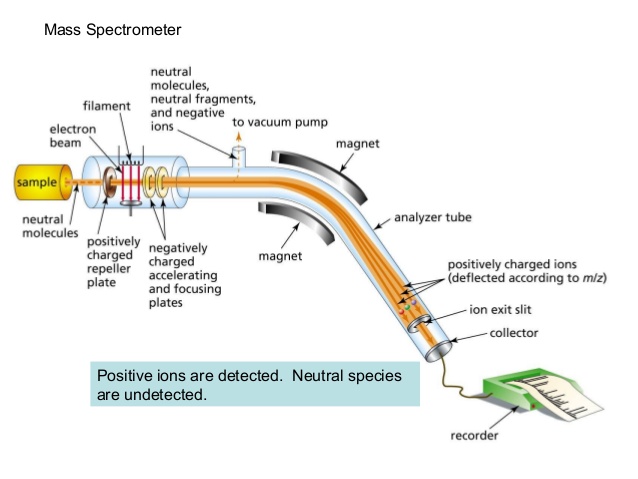
(Image from Wikipedia Commons)
This tube is curved, and the ions are deflected by a strong magnetic field. Ions of different mass to charge (m/z) ratios are deflected to a different extent, resulting in a ‘sorting’ of ions by mass (virtually all ions have charges of z = +1, so sorting by the mass to charge ratio is the same thing as sorting by mass). A detector at the end of the curved flight tube records and quantifies the sorted ions.
Looking at mass spectra
Below is typical output for an electron-ionization MS experiment (MS data in the section is derived from the Spectral Database for Organic Compounds, a free, web-based service provided by AIST in Japan.
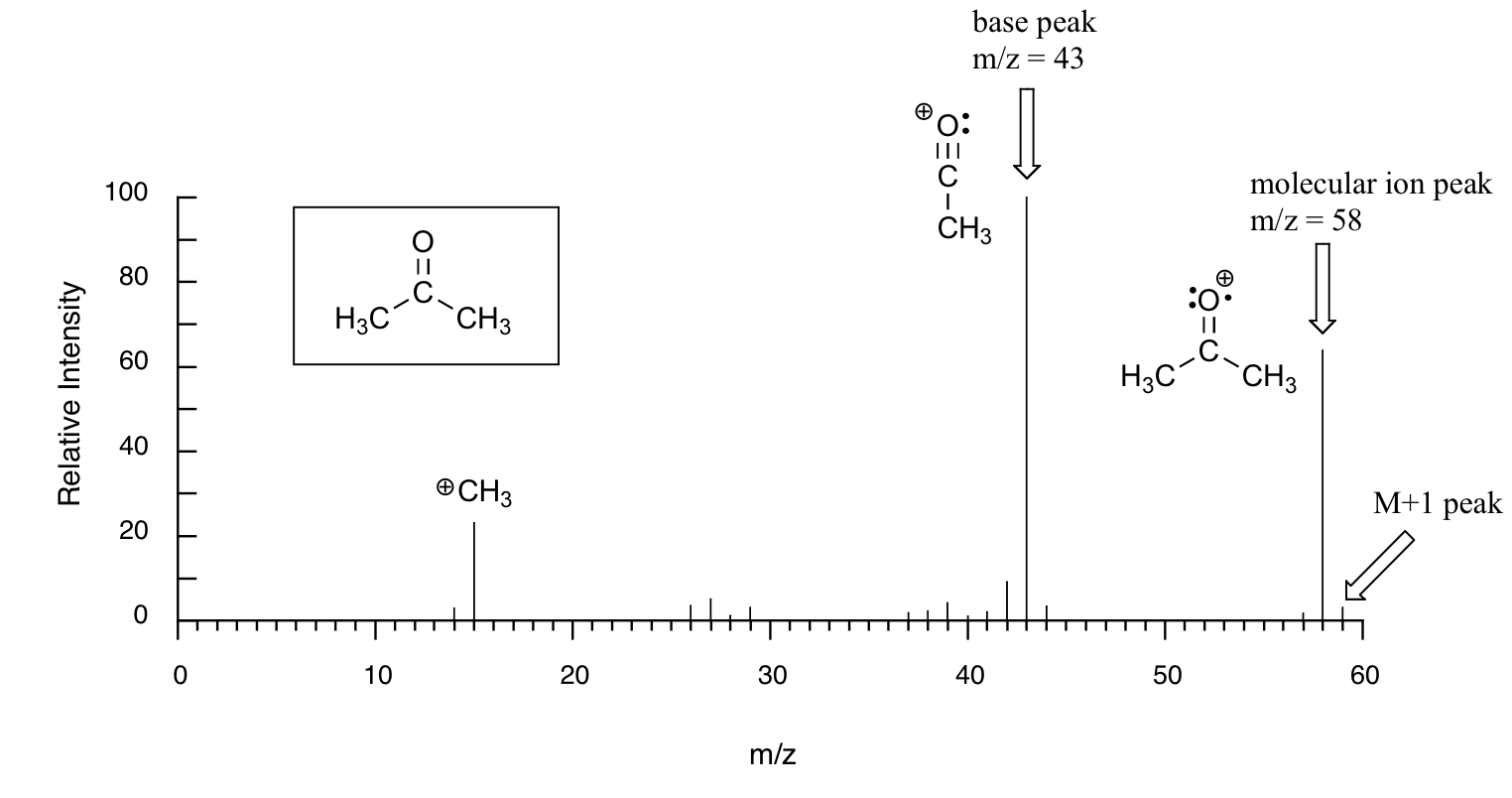
The sample is acetone. On the horizontal axis is the value for m/z (as we stated above, the charge z is almost always +1, so in practice this is the same as mass). On the vertical axis is the relative abundance of each ion detected. On this scale, the most abundant ion, called the base peak, is set to 100%, and all other peaks are recorded relative to this value. For acetone, the base peak is at m/z = 43 - we will discuss the formation of this fragment a bit later. The molecular weight of acetone is 58, so we can identify the peak at m/z = 58 as that corresponding to the molecular ion peak, or parent peak. Notice that there is a small peak at m/z = 59: this is referred to as the M+1 peak. How can there be an ion that has a greater mass than the molecular ion? Simple: a small fraction - about 1.1% - of all carbon atoms in nature are actually the 13C rather than the 12C isotope. The 13C isotope is, of course, heavier than 12C by 1 mass unit. In addition, about 0.015% of all hydrogen atoms are actually deuterium, the 2H isotope. So the M+1 peak represents those few acetone molecules in the sample which contained either a 13C or 2H.
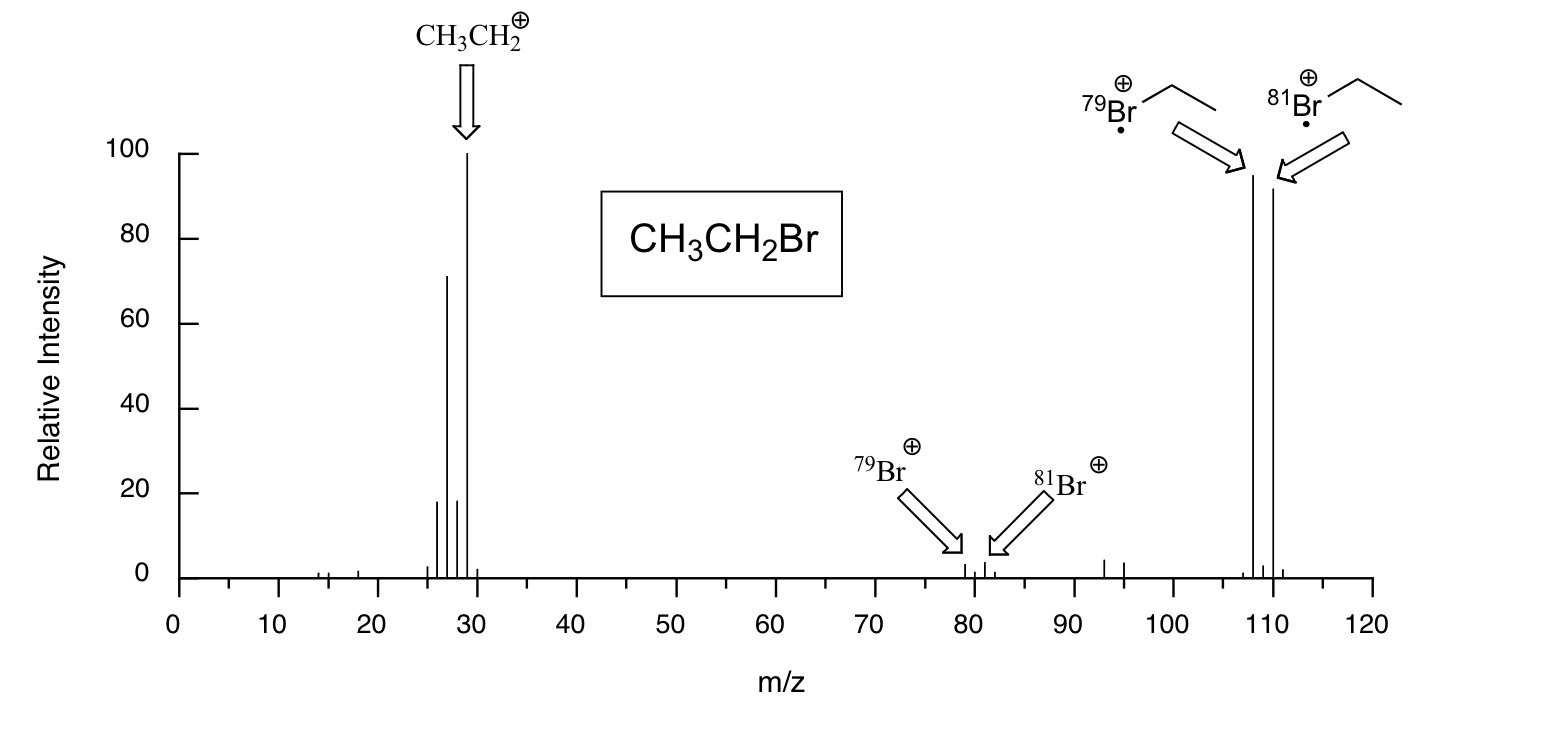
Molecules with lots of oxygen atoms sometimes show a small M+2 peak (2 m/z units greater than the parent peak) in their mass spectra, due to the presence of a small amount of 18O (the most abundant isotope of oxygen is 16O). Because there are two abundant isotopes of both chlorine (about 75% 35Cl and 25% 37Cl) and bromine (about 50% 79Br and 50% 81Br), chlorinated and brominated compounds have very large and recognizable M+2 peaks. Fragments containing both isotopes of Br can be seen in the mass spectrum of ethyl bromide:
Much of the utility in electron-ionization MS comes from the fact that the radical cations generated in the electron-bombardment process tend to fragment in predictable ways. Detailed analysis of the typical fragmentation patterns of different functional groups is beyond the scope of this text, but it is worthwhile to see a few representative examples, even if we don’t attempt to understand the exact process by which the fragmentation occurs. We saw, for example, that the base peak in the mass spectrum of acetone is m/z = 43. This is the result of cleavage at the ‘alpha’ position - in other words, at the carbon-carbon bond adjacent to the carbonyl. Alpha cleavage results in the formation of an acylium ion (which accounts for the base peak at m/z = 43) and a methyl radical, which is neutral and therefore not detected.
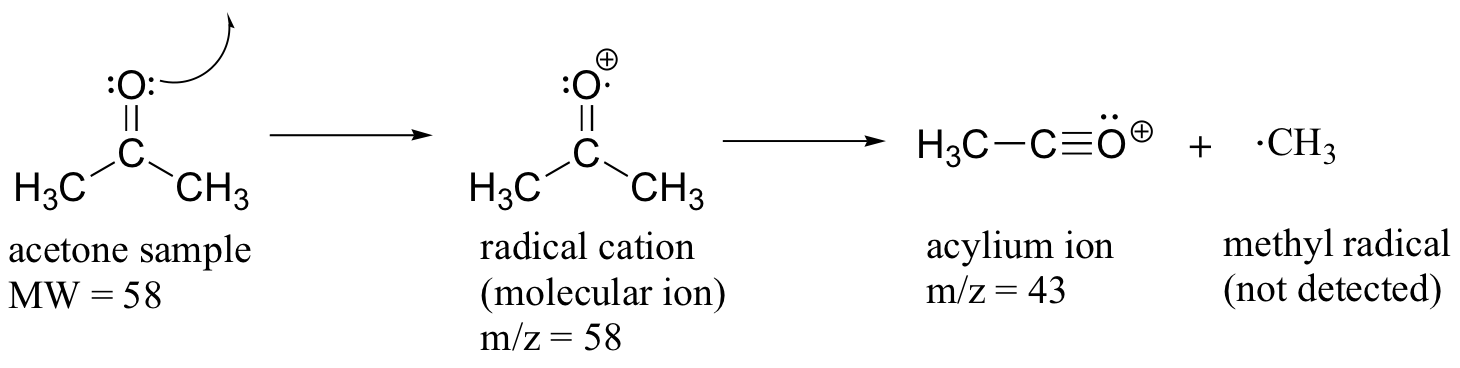
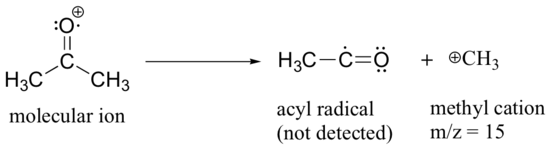
After the parent peak and the base peak, the next largest peak, at a relative abundance of 23%, is at m/z = 15. This, as you might expect, is the result of formation of a methyl cation, in addition to an acyl radical (which is neutral and not detected).
A common fragmentation pattern for larger carbonyl compounds is called the McLafferty rearrangement:

The mass spectrum of 2-hexanone shows a 'McLafferty fragment' at m/z = 58, while the propene fragment is not observed because it is a neutral species (remember, only cationic fragments are observed in MS). The base peak in this spectrum is again an acylium ion.

When alcohols are subjected to electron ionization MS, the molecular ion is highly unstable and thus a parent peak is often not detected. Often the base peak is from an ‘oxonium’ ion.
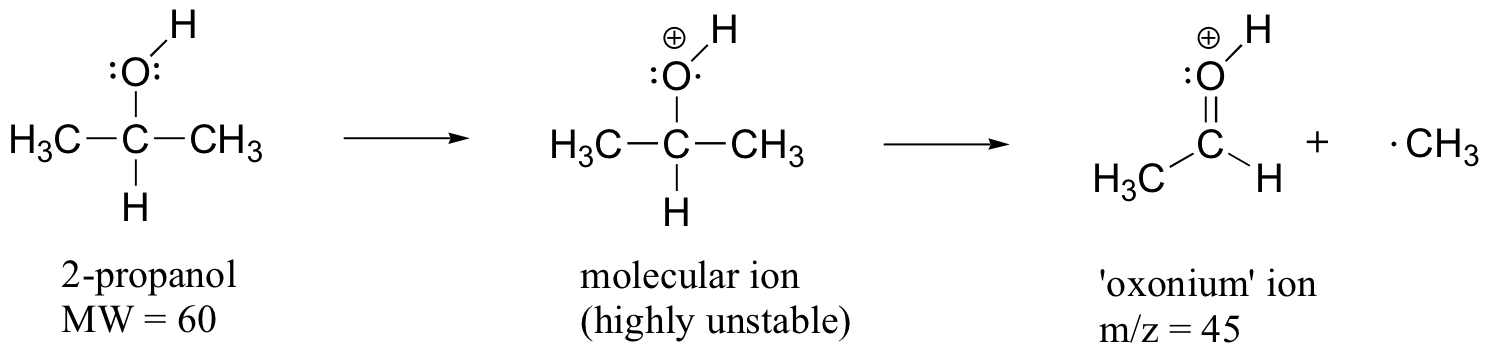
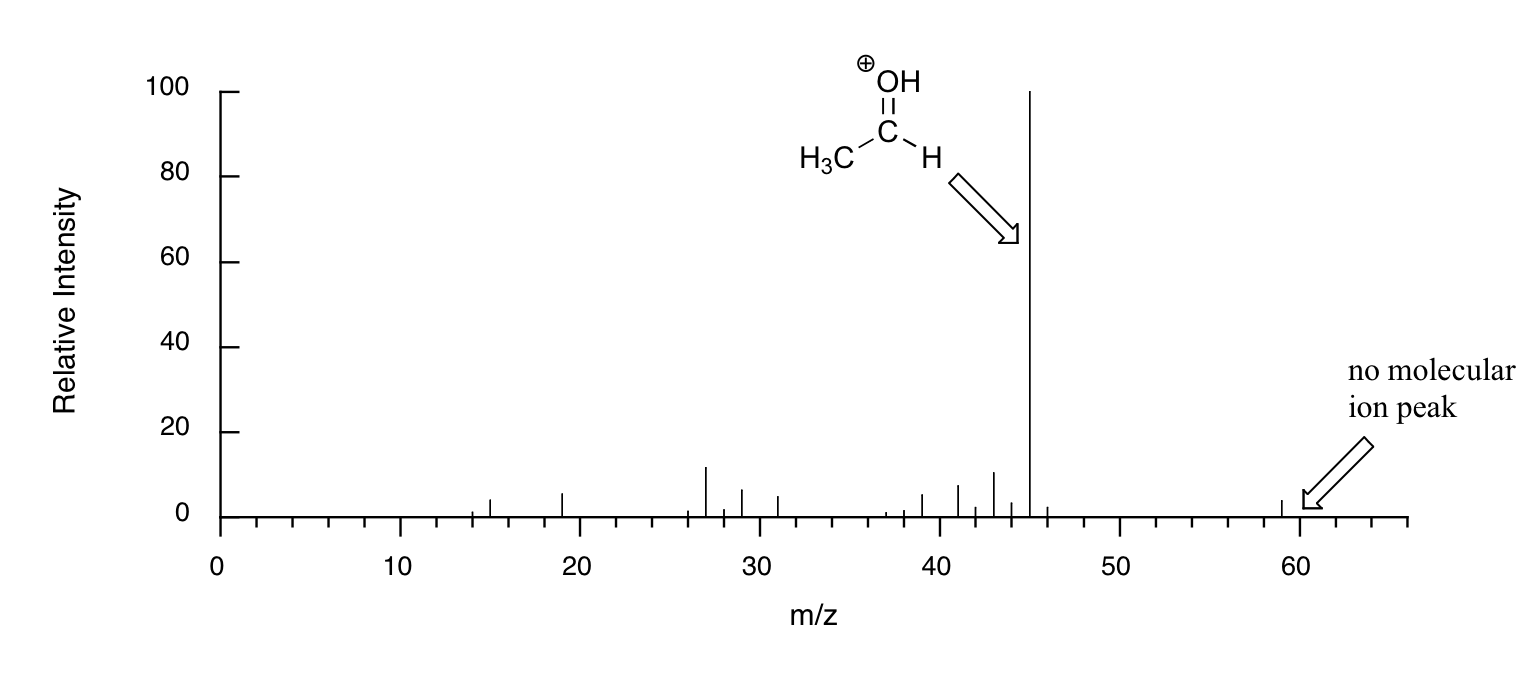
Other functional groups have predictable fragmentation patterns as well. By carefully analyzing the fragmentation information that a mass spectrum provides, a knowledgeable spectrometrist can often ‘put the puzzle together’ and make some very confident predictions about the structure of the starting sample. You can see many more actual examples of mass spectra in the Spectral Database for Organic Compounds
Using the fragmentation patterns for acetone as a guide, predict the signals that you would find in the mass spectra of:
a) 2-butanone; b) 3-hexanone; c) cyclopentanone
Predict some signals that you would expect to see in a mass spectrum of 2-chloropropane.
The mass spectrum of an aldehyde shows a parent peak at m/z = 58 and a base peak at m/z = 29. Propose a structure, and identify the two species whose m/z values were listed. (
Gas Chromatography - Mass Spectrometry
Quite often, mass spectrometry is used in conjunction with a separation technique called gas chromatography (GC). The combined GC-MS procedure is very useful when dealing with a sample that is a mixture of two or more different compounds, because the various compounds are separated from one another before being subjected individually to MS analysis. We will not go into the details of gas chromatography here, although if you are taking an organic laboratory course you might well get a chance to try your hand at GC, and you will almost certainly be exposed to the conceptually analogous techniques of thin layer and column chromatography. Suffice it to say that in GC, a very small amount of a liquid sample is vaporized, injected into a long, coiled metal column, and pushed though the column by helium gas. Along the way, different compounds in the sample stick to the walls of the column to different extents, and thus travel at different speeds and emerge separately from the end of the column. In GC-MS, each purified compound is sent directly from the end of GC column into the MS instrument, so in the end we get a separate mass spectrum for each of the compounds in the original mixed sample. Because a compound's MS spectrum is a very reliable and reproducible 'fingerprint', we can instruct the instrument to search an MS database and identify each compound in the sample.
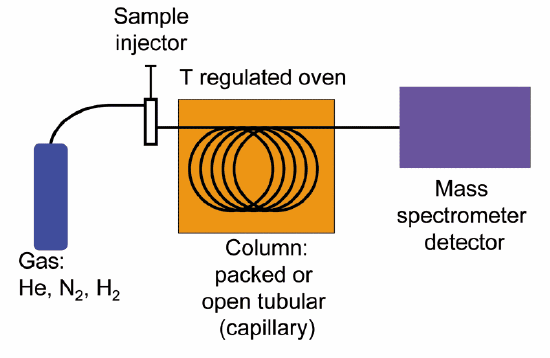
Gas chromatography-mass spectrometry (GC-MS) schematic
(Image from Wikipedia by K. Murray)
The extremely high sensitivity of modern GC-MS instrumentation makes it possible to detect and identify very small trace amounts of organic compounds. GC-MS is being used increasingly by environmental chemists to detect the presence of harmful organic contaminants in food and water samples. Airport security screeners also use high-speed GC-MS instruments to look for residue from bomb-making chemicals on checked luggage.
Mass spectrometry of proteins - applications in proteomics
Electron ionization mass spectrometry is generally not very useful for analyzing biomolecules: their high polarity makes it difficult to get them into the vapor phase, the first step in EIMS. Mass spectrometry of biomolecules has undergone a revolution over the past few decades, with many new ionization and separation techniques being developed. Generally, the strategy for biomolecule analysis involves soft ionization, in which much less energy (compared to techniques such as EIMS) is imparted to the molecule being analyzed during the ionization process. Usually, soft ionization involves adding protons rather than removing electrons: the cations formed in this way are significantly less energetic than the radical cations formed by removal of an electron. The result of soft ionization is that little or no fragmentation occurs, so the mass being measured is that of an intact molecule. Typically, large biomolecules are digested into smaller pieces using chemical or enzymatic methods, then their masses determined by 'soft' MS.
New developments in soft ionization MS technology have made it easier to detect and identify proteins that are present in very small quantities in biological samples. In electrospray ionization (ESI), the protein sample, in solution, is sprayed into a tube and the molecules are induced by an electric field to pick up extra protons from the solvent. Another common 'soft ionization' method is 'matrix-assisted laser desorption ionization' (MALDI). Here, the protein sample is adsorbed onto a solid matrix, and protonation is achieved with a laser.
Typically, both electrospray ionization and MALDI are used in conjunction with a time-of-flight (TOF) mass analyzer component.

The proteins are accelerated by an electrode through a column, and separation is achieved because lighter ions travel at greater velocity than heavier ions with the same overall charge. In this way, the many proteins in a complex biological sample (such as blood plasma, urine, etc.) can be separated and their individual masses determined very accurately. Modern protein MS is extremely sensitive – recently, scientists were even able to detect the presence of Tyrannosaurus rex protein in a fossilized skeleton! (Science 2007, 316, 277).
Soft ionization mass spectrometry has become in recent years an increasingly important tool in the field of proteomics. Traditionally, protein biochemists tend to study the structure and function of individual proteins. Proteomics researchers, in contrast, want to learn more about how large numbers of proteins in a living system interact with each other, and how they respond to changes in the state of the organism. One important subfield of proteomics is the search for protein 'biomarkers' for human disease: in other words, proteins which are present in greater quantities in the tissues of a sick person than in a healthy person. Detection in a healthy person of a known biomarker for a disease such as diabetes or cancer could provide doctors with an early warning that the patient may be especially susceptible to the disease, so that preventive measures could be taken to prevent or delay onset.
In a 2005 study, MALDI-TOF mass spectrometry was used to compare fluid samples from lung transplant recipients who had suffered from tissue rejection to samples from recipients who had not suffered rejection. Three peptides (short proteins) were found to be present at elevated levels specifically in the tissue rejection samples. It is hoped that these peptides might serve as biomarkers to identify patients who are at increased risk of rejecting their transplanted lungs. (Proteomics 2005, 5, 1705).