
3.2 Conformations of Alkanes
- Page ID
- 17023
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Conformational isomerism involves rotation about sigma bonds, and does not involve any differences in the connectivity or geometry of bonding. Two or more structures that are categorized as conformational isomers, or conformers, are really just two of the exact same molecule that differ only in terms of the angle about one or more sigma bonds.
Ethane Conformations
Although there are seven sigma bonds in the ethane molecule, rotation about the six carbon-hydrogen bonds does not result in any change in the shape of the molecule because the hydrogen atoms are essentially spherical. Rotation about the carbon-carbon bond, however, results in many different possible molecular conformations.
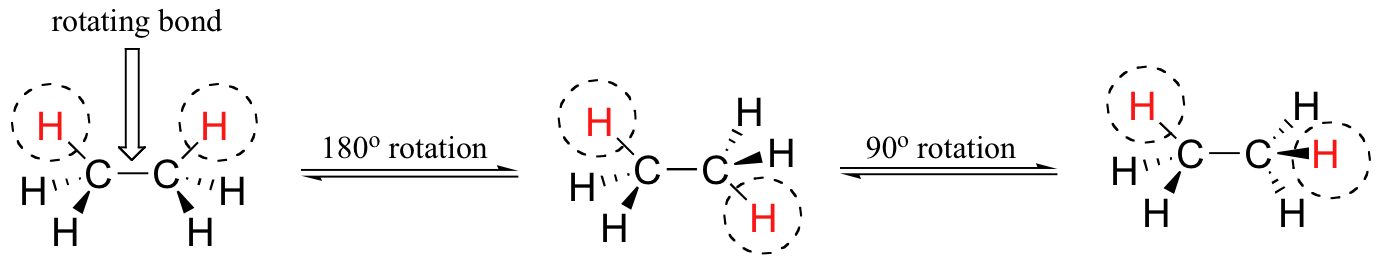
In order to better visualize these different conformations, it is convenient to use a drawing convention called the Newman projection. In a Newman projection, we look lengthwise down a specific bond of interest – in this case, the carbon-carbon bond in ethane. We depict the ‘front’ atom as a dot, and the ‘back’ atom as a larger circle.

The six carbon-hydrogen bonds are shown as solid lines protruding from the two carbons at 120°angles, which is what the actual tetrahedral geometry looks like when viewed from this perspective and flattened into two dimensions.
The lowest energy conformation of ethane, shown in the figure above, is called the ‘staggered’ conformation, in which all of the C-H bonds on the front carbon are positioned at dihedral angles of 60°relative to the C-H bonds on the back carbon. In this conformation, the distance between the bonds (and the electrons in them) is maximized.
If we now rotate the front CH3 group 60°clockwise, the molecule is in the highest energy ‘eclipsed' conformation, where the hydrogens on the front carbon are as close as possible to the hydrogens on the back carbon.
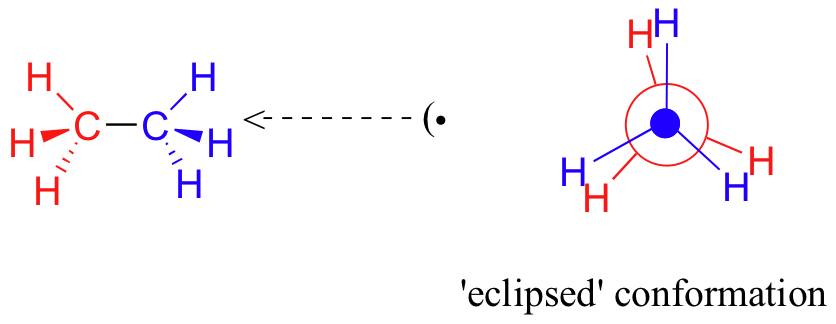
This is the highest energy conformation because of unfavorable interactions between the electrons in the front and back C-H bonds. The energy of the eclipsed conformation is approximately 3 kcal/mol higher than that of the staggered conformation.
Another 60°rotation returns the molecule to a second eclipsed conformation. This process can be continued all around the 360°circle, with three possible eclipsed conformations and three staggered conformations, in addition to an infinite number of variations in between.
The carbon-carbon bond is not completely free to rotate – there is indeed a small, 3 kcal/mol barrier to rotation that must be overcome for the bond to rotate from one staggered conformation to another. This rotational barrier is not high enough to prevent constant rotation except at extremely cold temperatures. However, at any given moment the molecule is more likely to be in a staggered conformation - one of the rotational ‘energy valleys’ - than in any other state.
The potential energy associated with the various conformations of ethane varies with the dihedral angle of the bonds, as shown below.
Although the conformers of ethane are in rapid equilibrium with each other, the 3 kcal/mol energy difference leads to a substantial preponderance of staggered conformers (> 99.9%) at any given time.
The animation below illustrates the relationship between ethane's potential energy and its dihedral angle
Butane Conformations
The hydrocarbon butane has a larger and more complex set of conformations associated with its constitution than does ethane. Of particular interest and importance are the conformations produced by rotation about the central carbon-carbon bond. Among these we shall focus on two staggered conformers (A & C) and two eclipsed conformers (B & D), shown below in several stereo-representations.
As in the case of ethane, the staggered conformers are more stable than the eclipsed conformers by 2.8 to 4.5 kcal/mol. Since the staggered conformers represent the chief components of a butane sample they have been given the identifying prefix designations anti for A and gauche for C.
The following diagram illustrates the change in potential energy that occurs with rotation about the C2–C3 bond.
Here is a summary of the most important aspects of conformational stereoisomerism:
(i) Most conformational interconversions in simple molecules occur rapidly at room temperature. Consequently, isolation of pure conformers is usually not possible.
(ii) Specific conformers require special nomenclature terms such as staggered, eclipsed, gauche and anti when they are designated.
(iii) Specific conformers may also be designated by dihedral angles. In the butane conformers shown above, the dihedral angles formed by the two methyl groups about the central double bond are: A 180º, B 120º, C 60º & D 0º.
(iv) Staggered conformations about carbon-carbon single bonds are more stable (have a lower potential energy) than the corresponding eclipsed conformations. The higher energy of eclipsed bonds is known as eclipsing strain.
(v) In butane the gauche-conformer is less stable than the anti-conformer by about 0.9 kcal/mol. This is due to a crowding of the two methyl groups in the gauche structure, and is called steric strain or steric hindrance.
Further Reading
Slide presentation on alkane conformations
Conformational Analysis Problems
Conformation tutorial questions
Conformation of Ethane
Carey 4th Edition On-Line Activity
Conformations of simple alkanes
Khan Academy
Newman Projections
MasterOrganicChemistry
Carey 4th Edition On-Line Activity
Khan Academy
Leah4Sci
Web Pages
Videos
Tutorial
How to Draw Newman Projections
Long Newman projection tutorial
How to draw Newman projections
Switching between Newman projections and dash-wedge drawings
Other tools