
17.03.1: Origins of Acidity of Carboxylic Acids
- Page ID
- 26149
Carboxylic acids, containing a carboxyl group (-COOH), are the most acidic class of standard organic molecules. Carboxylic acids generally have pKas in the range of 3 - 5, and therefore are weaker acids than hydronium ion (H3O+), but they are stronger acids than other organic acids, such as alcohols (16 - 20), aldehydes and ketones (18 - 22), alkynes (25), benzene (35) or alkanes (50).
Because they are relatively strong acids, the reactivity of carboxylic acids is dominated by acid-base chemistry. Therefore, reactions with most strong nucleophiles, which are also generally strong bases, occurs by proton-transfer, and not by nucleophilic addition, as would occur with aldehydes, ketones and carboxylic acid derivatives. The types of nucleophiles that react by carbonyl addition with aldehydes/ketones but deprotonate carboxylic acids includes organometallic reagents (Grignard and organolithium reagents), acetylides, nitriles and amines. The most important thing to remember is that "Carboxylic Acids are Acids."
Origins of the Enhanced Acidity of Carboxylic Acids
Resonance
The common explanation for why carboxylic acids are more acidic than other molecules (such as alcohols) is that resonance delocalization of charge stabilizes the conjugate base anion relative to the reactant acid. Examples can be seen on the chemwiki pages, elsewhere on-line (here and here, for example) and is the standard text-book explanation.
Inductive effects
During the last 25 - 30 years, the resonance explanation for the enhanced acidity of carboxylic acids has been questioned. In a seminal study, Rablen asked the simple question, "...why is CH3CO2H called acetic acid, and not "acetic base."1 As he argued, if resonance delocalization of charge was really that important, carboxylic acids should be strong basis, as well as being strong acids. Upon protonation, the charge can also be delocalized by resonance.
However, carboxylic acids are, in fact, less basic than simple ketones or aldehydes.
Moreover, although carbonic acid (HO-COOH) is more acidic than acetic acid, it is less basic. From a resonance perspective, this is surprising because there is no change in the number of resonance structures available in the bicarbonate anion, protonated carbonic acid provides an additional resonance structure to delocalize charge.
An explanation that does account for these observations is that it is not resonance that is important, it is inductive effects of the carbonyl, or, more specifically, the inductive effects of C-O bonds, that are more important.
Most textbooks do discuss the role of inductive effects on carboxylic acid acidity. However, they focus on the inductive effects of substituents added to the carbonyl. Thus, trichloroacetic acid (pKa = 0.9) is more acidic than dichloroacetic acid (pKa = 1.3) which more acidic than chloroacetic acid (pKa = 2.9) which is more acidic than acetic acid (pKa = 4.7). This trend is explained as due to the increasiing inductive stabilization of the carboxylate anion due to the addition of chlorine substituents.
The carbonyl group contains a highly polarized C-O pi bond, such that the carbon of the carbonyl is electron deficient. Consequently, upon deprotonation, the oxo-anion is next to an electron deficient carbon.
Irrespective of any resonance, the acidity of oxy-acids are increased by the addition of inductively withdrawing groups. For example, as with the series of chlorinated acetic acids, the acidities of alcohols are increased with the addiiton of inductively withdrawing groups.
| Alcohol | pKa |
| Ethanol | 16 |
| CF3CH2-OH | 12 |
| (CF3)3C-OH | 5.4 |
| CH3C(O)-OH | 4.7 |
Given the carbonyl, with an electron-deficient carbon, is inductively withdrawing, perhaps it is the inductive effect that stabilizes the negative ion, and not resonance?
Many studies have considered this issue, and the common conclusion is that the enhanced acidity of carboxylic acids results predominantly from inductive effects, although resonance also plays a minor role. Depending on the model, the acidity enhancement is generally found to be 70 - 80% due to inductive effects, and 20 - 30% due to resonance.
Inductive effect of a carbonyl group
The pKas of fluorinated ethanols suggests that the effect of a carbonyl group on the acidity of is about the same as that of a fluorinated-tert-butyl group. "Group electronegativities" refer to the effective negativities of substituent groups, as opposed to individual atoms.2 The effective group electronegativity of the carbonyl is evident in effects that do not depend on resonance. For example, H NMR shifts for protons on carbon next to a carbonyl. Shifts of 2.1 - 2.2 ppm indicate that the carbonyl leads to deshielding of the protons, consistent with what is observed for protons on carbon connected to electronegative elements. The effect of the carbonyl on chemical shift is similar to that created by an iodine, and is slightly larger than that predicted for a CF3 group. Therefore, the inductive effect of a carbonyl should be similar to that for iodine or CF3. Just as there is no need to invoke resonance to account for the acidity of IOH or CF3OH, it should not be necessary to invoke resonance to account for the acidity of carboxylic acids.
Limitation of the Resonance Explanation
As noted above, the best estimates are that 70 - 80% of the enhanced acidity of carboxylic acids is due to inductive effects and only 20 - 30% can be attributed to resonance stabilization of the anion. Why does resonance contribute so little? It seems to make sense - clearly the anion is resonance delocalized. Shouldn't that result in energy stabilization?
The problem with the resonance explanation was initially pointed out by Siggel and Thomas,3 in the work that started the whole process of reconsidering the origins of the acidity in carboxylic acids. Upon measuring ionization energies of core (1s) electrons on carbon and oxygen, they discovered that there was little change in the extent of electron delocalization in the carboxyl group upon deprotonation. Therefore, the premise that there is more favorable delocalization in the anion is not as true as it would appear. Much of the delocalization that is found in the anion is already present in the neutral carboxylic acid.
Consequently, the enhanced acidity of acetic acid can be viewed as due to enhancement of the neutral carboxylic acid, which has extensive positive charge on the proton due to the electron withdrawing capability of the carbonyl. There is no need to invoke the properties of the anion.
Applications
Looking at the resonance within the reactant can be used to assess other systems as well. For example, amides are well-recognized to have extensive resonance interaction in the neutral, to the extent that the nitrogen atom adopts a planar geometry and sp2 hybridization.
Yet, the acidity difference between acetamide and ethyl amine is greater than the acidity difference between acetic acid and ethanol.4
Also, if the amide is constrained to a geometry where the protons on nitrogen are constrained to be out of the plane, such that no resonance interaction can occur, the acidity enhancement is calculated to be 150 kJ/mol, so just as large as when resonance is present. Therefore, there is no indication that resonance has any role in enhancing the acidity of amides.
In contrast, acetone is case where there is no resonance delocalization in the reactant. In that case, the acidity enhancement is 210 kJ/mol, compared to propane.4
However, if the enolate is twisted, such that resonance can not occur, the enhancement is only 50 kJ/mol. This is the component that can be attributed to the inductive effect of the carbonyl. The rest of the enhancement can be attributed to resonance stabilization
The rest of the enhancement can be attributed to resonance stabilization of the anion. In particular, the anion is highly stabilized by the ability to distribute negative charge on to the carbonyl oxygen.
Problem
1. Vitamin C is a relatively strong acid, with a pKa of 4.10.5 The structure of vitamin C is shown below.
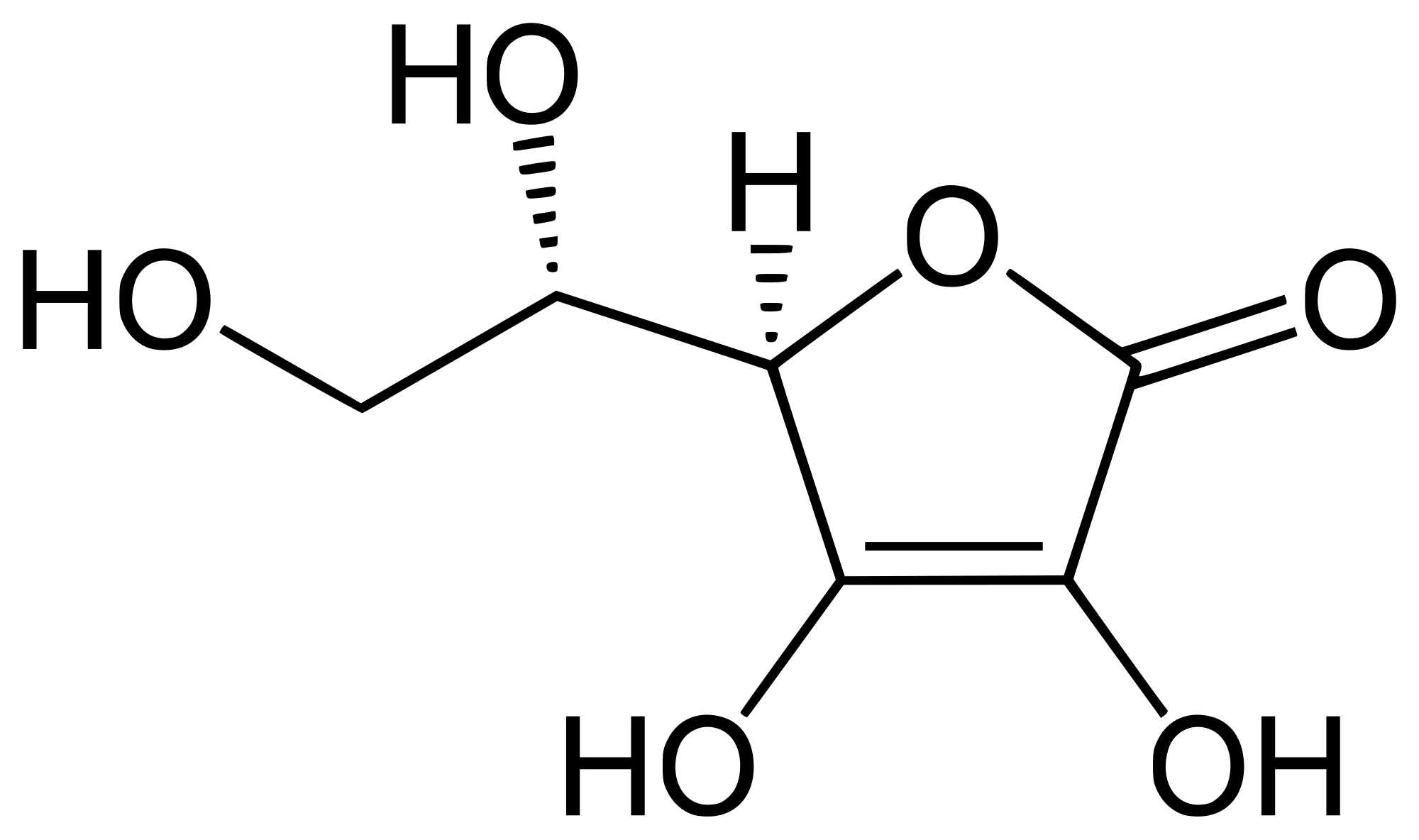
Which is the most acidic proton, and explain why it is a strong acid.
Answers
1. The aliphatic protons are not very acidic, so ignore them. There are two ways to explain the acidity of Vitamin C.
Standard Answer
The standard answer for determining the acidity of Vitamin C is to compare the anions obtained by deprotonation.
When deprotonation occurs at the 4-position, there is an extra, good resonance structure that can be drawn, and so the charge is delocalized. It works, and gives the correct prediction for which proton is most acidic.
Alternate Answer
We can also answer the question using the explanation similar to that used for carboxylic acids in this article. Upon polarizing the carbonyl, it is possible to draw resonance delocalizaiton in the neutral acid.
By examiniation of the structure on the right, it is immediately apparent that the most acidic position in the molecule is at position 4, where there is extensive positive charge character at the proton.
References
1. Rablen, P. R. J. Am. Chem. Soc. 2000, 122, 357-368.
2. Wells, P. R. Prog. Phys. Org. Chem., 1968, 6, 111.
3. Siggel, M. R.; Thomas, T. D. J. Am. Chem. Soc. 1986, 108, 4360-4363