
Organic Acids
- Page ID
- 3654

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)This page explains the acidity of simple organic acids and looks at the factors which affect their relative strengths.
Organic acids as weak acids
For the purposes of this topic, we are going to take the definition of an acid as "a substance which donates hydrogen ions (protons) to other things". We are going to get a measure of this by looking at how easily the acids release hydrogen ions to water molecules when they are in solution in water.
An acid in solution sets up this equilibrium:
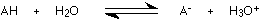
A hydroxonium ion is formed together with the anion (negative ion) from the acid.
This equilibrium is sometimes simplified by leaving out the water to emphasise the ionisation of the acid.
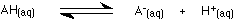
If you write it like this, you must include the state symbols - "(aq)". Writing H+(aq) implies that the hydrogen ion is attached to a water molecule as H3O+. Hydrogen ions are always attached to something during chemical reactions.
The organic acids are weak in the sense that this ionisation is very incomplete. At any one time, most of the acid will be present in the solution as un-ionised molecules. For example, in the case of dilute ethanoic acid, the solution contains about 99% of ethanoic acid molecules - at any instant, only about 1% have actually ionised. The position of equilibrium therefore lies well to the left.
Comparing the strengths of weak acids
The strengths of weak acids are measured on the pKa scale. The smaller the number on this scale, the stronger the acid is.
Three of the compounds we shall be looking at, together with their pKa values are:
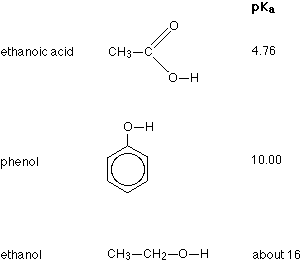
Remember - the smaller the number the stronger the acid. Comparing the other two to ethanoic acid, you will see that phenol is very much weaker with a pKa of 10.00, and ethanol is so weak with a pKa of about 16 that it hardly counts as acidic at all!
Why are these acids acidic?
In each case, the same bond gets broken - the bond between the hydrogen and oxygen in an -OH group. Writing the rest of the molecule as "X":
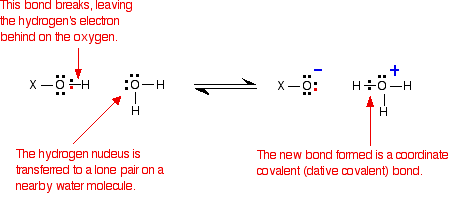
So . . . if the same bond is being broken in each case, why do these three compounds have such widely different acid strengths?
Differences in acid strengths between carboxylic acids, phenols and alcohols
Two of the factors which influence the ionization of an acid are:
- the strength of the bond being broken,
- the stability of the ions being formed.
In these cases, you seem to be breaking the same oxygen-hydrogen bond each time, and so you might expect the strengths to be similar. The most important factor in determining the relative acid strengths of these molecules is the nature of the ions formed. You always get a hydroxonium ion - so that's constant - but the nature of the anion (the negative ion) varies markedly from case to case.
Ethanoic acid has the structure:
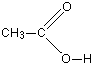
The acidic hydrogen is the one attached to the oxygen. When ethanoic acid ionises it forms the ethanoate ion, CH3COO-.
You might reasonably suppose that the structure of the ethanoate ion was as below, but measurements of bond lengths show that the two carbon-oxygen bonds are identical and somewhere in length between a single and a double bond.

To understand why this is, you have to look in some detail at the bonding in the ethanoate ion. Like any other double bond, a carbon-oxygen double bond is made up of two different parts. One electron pair is found on the line between the two nuclei - this is known as a sigma bond. The other electron pair is found above and below the plane of the molecule in a pi bond. Pi bonds are made by sideways overlap between p orbitals on the carbon and the oxygen.
In an ethanoate ion, one of the lone pairs on the negative oxygen ends up almost parallel to these p orbitals, and overlaps with them
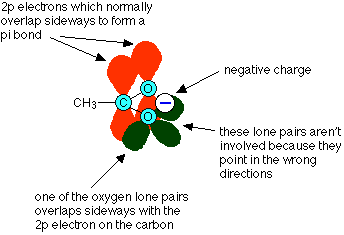
This leads to a delocalised pi system over the whole of the -COO- group, rather like that in benzene.
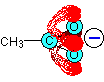
All the oxygen lone pairs have been left out of this diagram to avoid confusion. Because the oxygens are more electronegative than the carbon, the delocalised system is heavily distorted so that the electrons spend much more time in the region of the oxygen atoms.
So where is the negative charge in all this? It has been spread around over the whole of the -COO- group, but with the greatest chance of finding it in the region of the two oxygen atoms. Ethanoate ions can be drawn simply as:
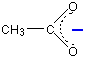
The dotted line represents the delocalisation. The negative charge is written centrally on that end of the molecule to show that it isn't localised on one of the oxygen atoms. The more you can spread charge around, the more stable an ion becomes. In this case, if you delocalise the negative charge over several atoms, it is going to be much less attractive to hydrogen ions - and so you are less likely to re-form the ethanoic acid.
Phenols have an -OH group attached directly to a benzene ring. Phenol itself is the simplest of these with nothing else attached to the ring apart from the -OH group.
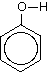
When the hydrogen-oxygen bond in phenol breaks, you get a phenoxide ion, C6H5O-.
Delocalisation also occurs in this ion. This time, one of the lone pairs on the oxygen atom overlaps with the delocalised electrons on the benzene ring.
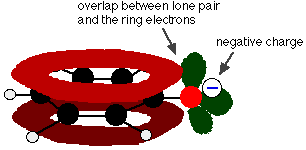
This overlap leads to a delocalisation which extends from the ring out over the oxygen atom. As a result, the negative charge is no longer entirely localised on the oxygen, but is spread out around the whole ion.
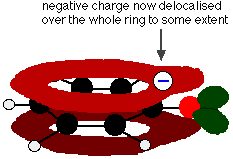
Why then is phenol a much weaker acid than ethanoic acid?
Think about the ethanoate ion again. If there wasn't any delocalisation, the charge would all be on one of the oxygen atoms, like this:
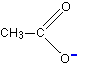
But the delocalisation spreads this charge over the whole of the COO group. Because oxygen is more electronegative than carbon, you can think of most of the charge being shared between the two oxygens (shown by the heavy red shading in this diagram).
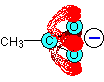
If there wasn't any delocalisation, one of the oxygens would have a full charge which would be very attractive towards hydrogen ions. With delocalisation, that charge is spread over two oxygen atoms, and neither will be as attractive to a hydrogen ion as if one of the oxygens carried the whole charge.
That means that the ethanoate ion won't take up a hydrogen ion as easily as it would if there wasn't any delocalisation. Because some of it stays ionised, the formation of the hydrogen ions means that it is acidic.
In the phenoxide ion, the single oxygen atom is still the most electronegative thing present, and the delocalised system will be heavily distorted towards it. That still leaves the oxygen atom with most of its negative charge.
What delocalisation there is makes the phenoxide ion more stable than it would otherwise be, and so phenol is acidic to an extent.
However, the delocalisation hasn't shared the charge around very effectively. There is still lots of negative charge around the oxygen to which hydrogen ions will be attracted - and so the phenol will readily re-form. Phenol is therefore only very weakly acidic.
Ethanol, CH3CH2OH, is so weakly acidic that you would hardly count it as acidic at all. If the hydrogen-oxygen bond breaks to release a hydrogen ion, an ethoxide ion is formed:
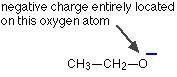
This has nothing at all going for it. There is no way of delocalising the negative charge, which remains firmly on the oxygen atom. That intense negative charge will be highly attractive towards hydrogen ions, and so the ethanol will instantly re-form.
Since ethanol is very poor at losing hydrogen ions, it is hardly acidic at all.
Variations in acid strengths between different carboxylic acids
You might think that all carboxylic acids would have the same strength because each depends on the delocalization of the negative charge around the -COO- group to make the anion more stable, and so more reluctant to re-combine with a hydrogen ion.
In fact, the carboxylic acids have widely different acidities. One obvious difference is between methanoic acid, HCOOH, and the other simple carboxylic acids:
| pKa | |
| HCOOH | 3.75 |
| CH3COOH | 4.76 |
| CH3CH2COOH | 4.87 |
| CH3CH2CH2COOH | 4.82 |
Remember that the higher the value for pKa, the weaker the acid is.
Why is ethanoic acid weaker than methanoic acid? It again depends on the stability of the anions formed - on how much it is possible to delocalise the negative charge. The less the charge is delocalised, the less stable the ion, and the weaker the acid.
The methanoate ion (from methanoic acid) is:
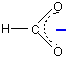
The only difference between this and the ethanoate ion is the presence of the CH3 group in the ethanoate.
But that's important! Alkyl groups have a tendency to "push" electrons away from themselves. That means that there will be a small amount of extra negative charge built up on the -COO- group. Any build-up of charge will make the ion less stable, and more attractive to hydrogen ions.
Ethanoic acid is therefore weaker than methanoic acid, because it will re-form more easily from its ions.
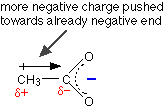
The other alkyl groups have "electron-pushing" effects very similar to the methyl group, and so the strengths of propanoic acid and butanoic acid are very similar to ethanoic acid. The acids can be strengthened by pulling charge away from the -COO- end. You can do this by attaching electronegative atoms like chlorine to the chain.
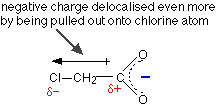
As the next table shows, the more chlorines you can attach the better:
| pKa | |
| CH3COOH | 4.76 |
| CH2ClCOOH | 2.86 |
| CHCl2COOH | 1.29 |
| CCl3COOH | 0.65 |
Trichloroethanoic acid is quite a strong acid.
Attaching different halogens also makes a difference. Fluorine is the most electronegative and so you would expect it to be most successful at pulling charge away from the -COO- end and so strengthening the acid.
| pKa | |
| CH2FCOOH | 2.66 |
| CH2ClCOOH | 2.86 |
| CH2BrCOOH | 2.90 |
| CH2ICOOH | 3.17 |
The effect is there, but isn't as great as you might expect.
Finally, notice that the effect falls off quite quickly as the attached halogen gets further away from the -COO- end. Here is what happens if you move a chlorine atom along the chain in butanoic acid.
| pKa | |
| CH3CH2CH2COOH | 4.82 |
| CH3CH2CHClCOOH | 2.84 |
| CH3CHClCH2COOH | 4.06 |
| CH2ClCH2CH2COOH | 4.52 |
The chlorine is effective at withdrawing charge when it is next-door to the -COO- group, and much less so as it gets even one carbon further away.