15.2: Regioselectivity in Electrophilic Aromatic Substitution
- Page ID
- 367990
We now have a good list of deactivating and activating groups that can be attached to the benzene ring, but two questions remain:
1. Are there other groups to add to this list? YES!
2. What do any of the groups in the reactivity series do to reactivity? A LOT!
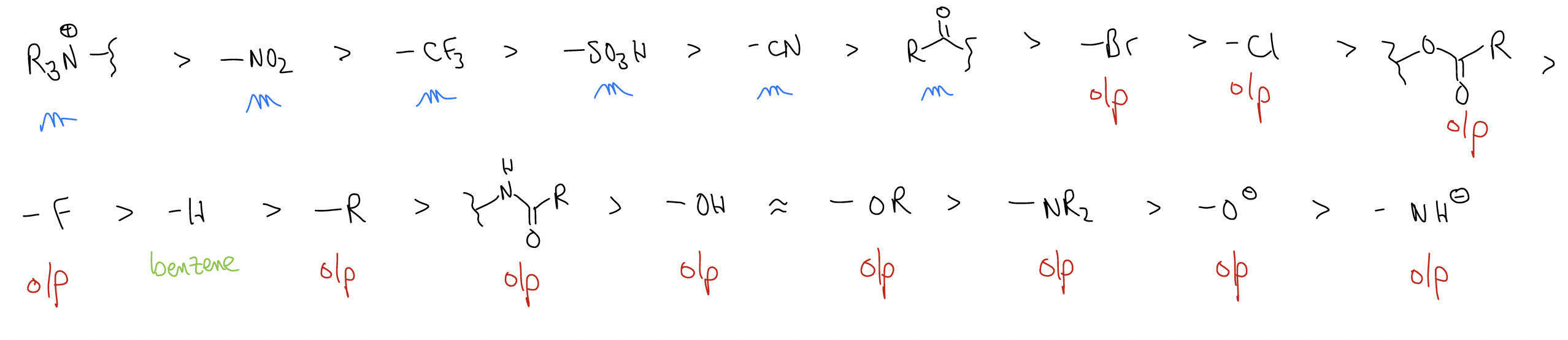
Here is a longer list of substituents that may be directly attached to a benzene ring:
Why is something deactivating?
- induction through the \(σ\)-network; ex. Br, Cl, F are all electronegative
- resonance delocalization pulls electron density out of the ring; ex. NO2, CN, R(C=O)
Why is something activating?
- a lone pair or \(π\) bond is directly aligned with the \(π\) system and can participate in resonance
- a \(σ\) bond is directly aligned with the \(π\) system and can participate in \(σ\)-donation
None of this tells us anything about where an electrophile will substitute if these groups are already on the ring. What is the ortho-, meta-, or para-directing ability of these groups? In other words, what is the regiochemistry of these reactions?
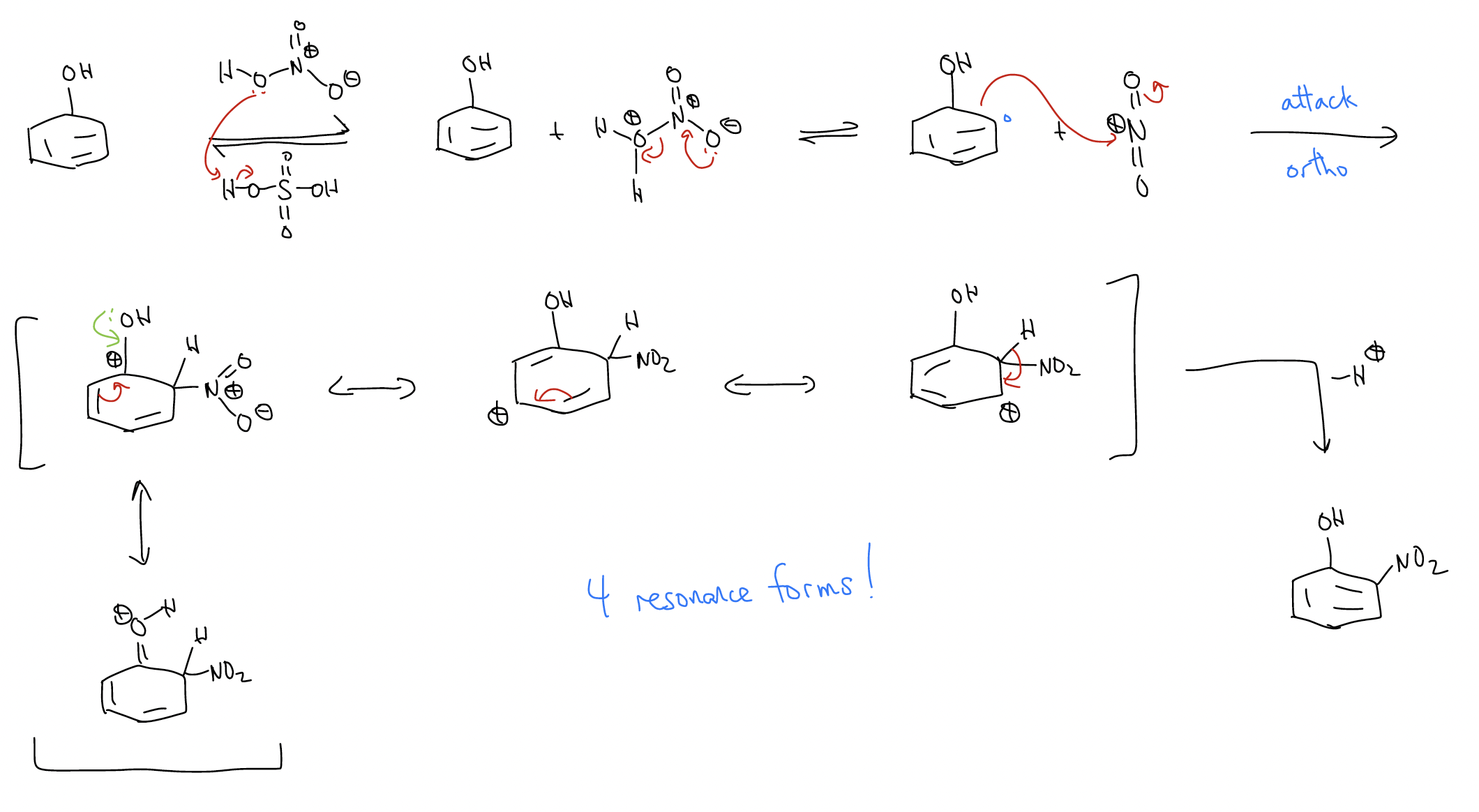
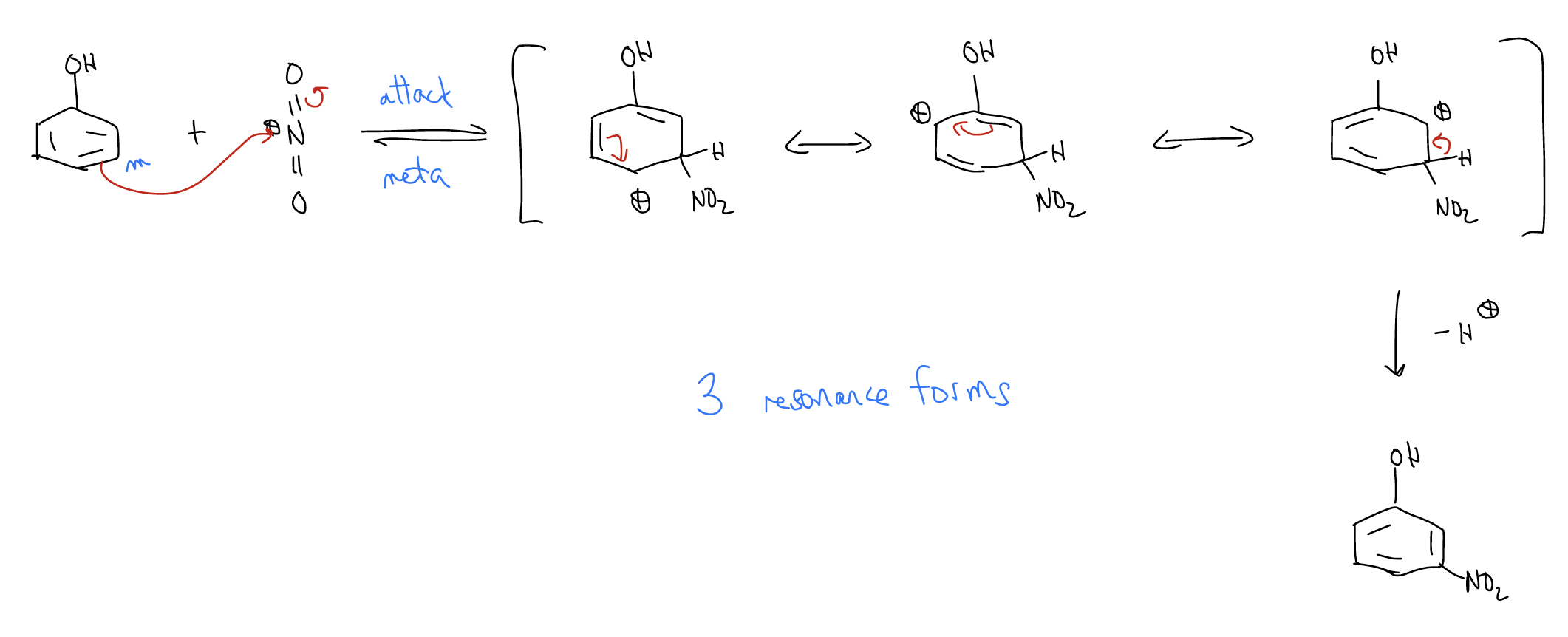
Let’s take the nitration of phenol. One two of a possible three monosubstituted products form (ortho and para). Why not meta? Well, let’s look at the mechanism:
a) ortho-nitration
b) meta-nitration
c) para-nitration
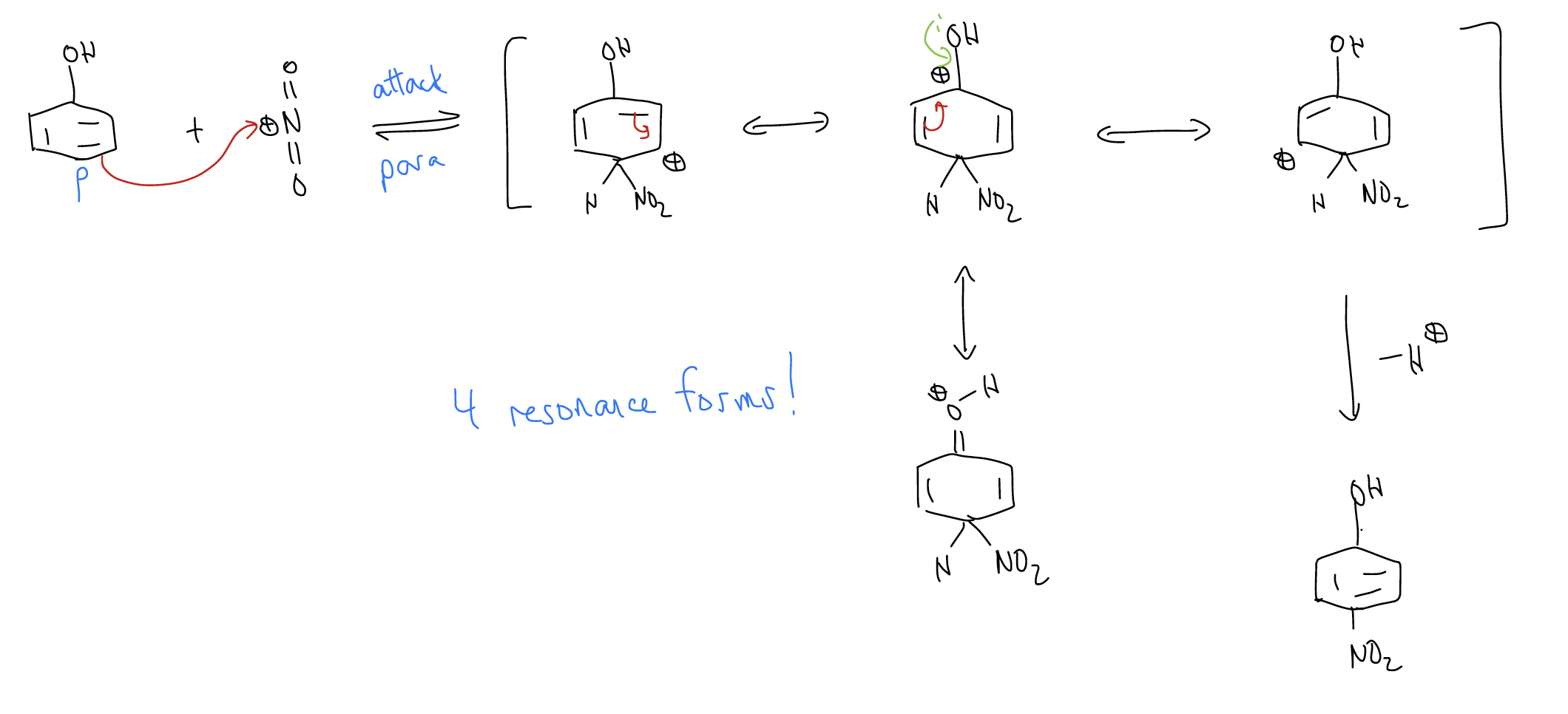
So, if we observed only the ortho- and para-nitrophenol products, can we devise a logical mechanistic reason for the regiochemistry? Well, since the arenium ion is more stabilized when the nitro group ads to the ortho- and para-positions, these two arenium ions will form faster. The activation energy needed to break aromaticity is smaller. Another way of looking at this is through electron density, which we can see from the 1H and 13C NMR chemical shifts.
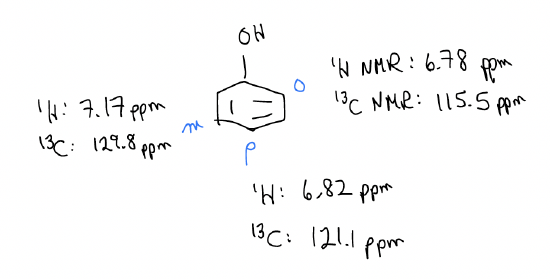
The NMR data tell you that the ortho- and para-positions have greater electron density. This should make sense because we can draw dipolar resonance forms placing negative charge at those positions.
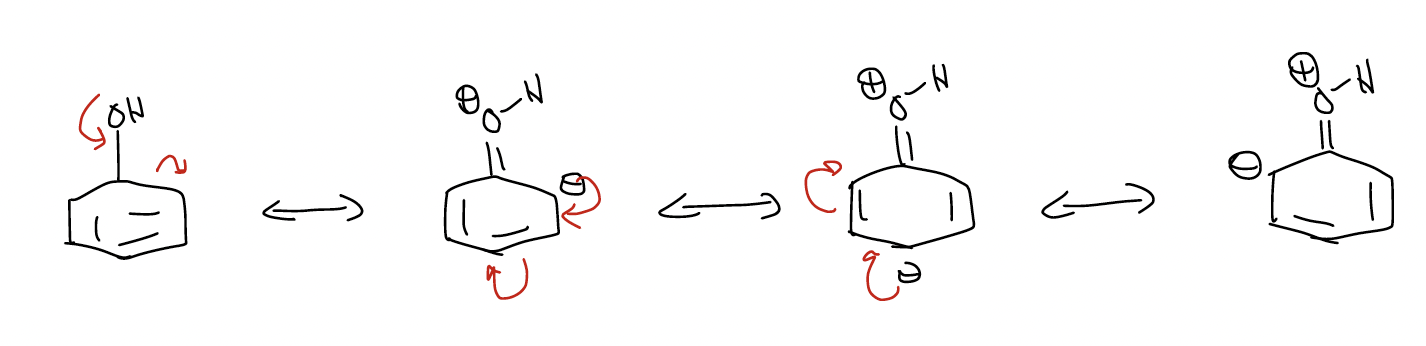
In other words, the ortho- and para-positions have greater \(δ^{–}\) charge. Since rate is controlled by electrostatic interactions, ortho- and para-substitution occurs exclusively.
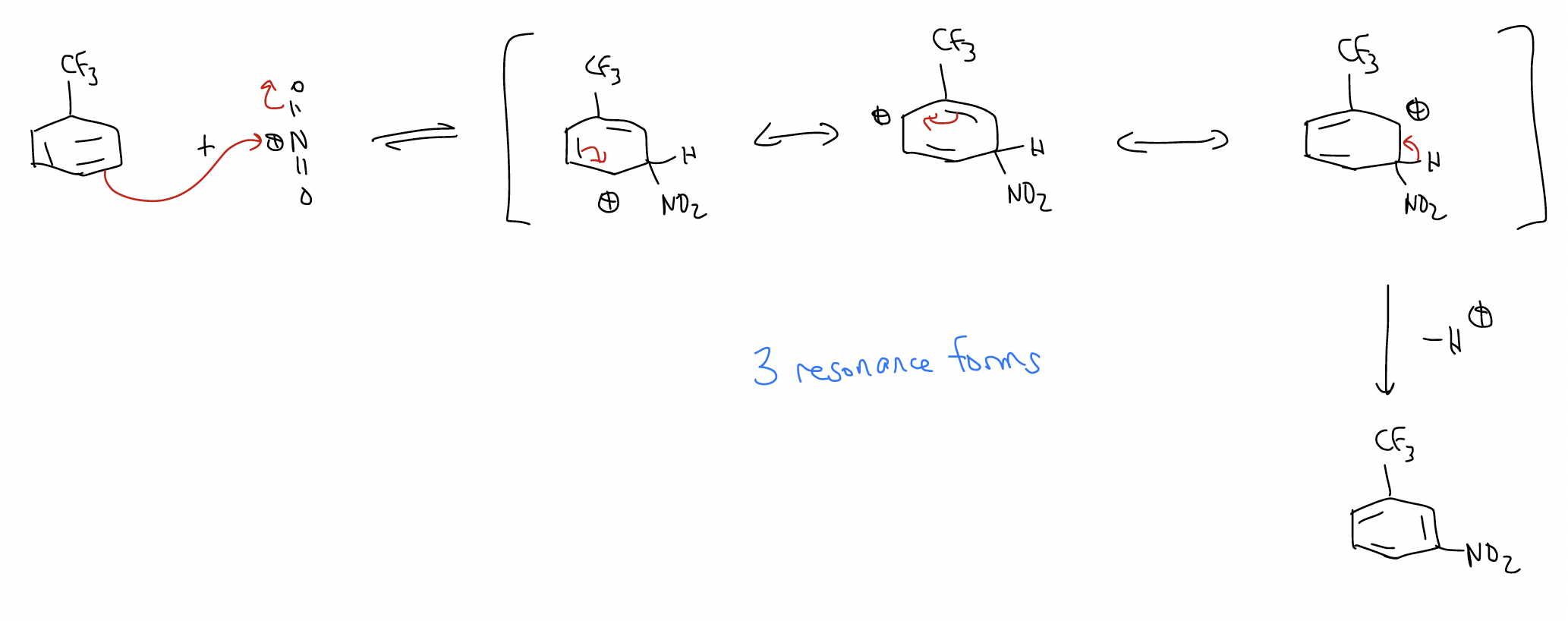
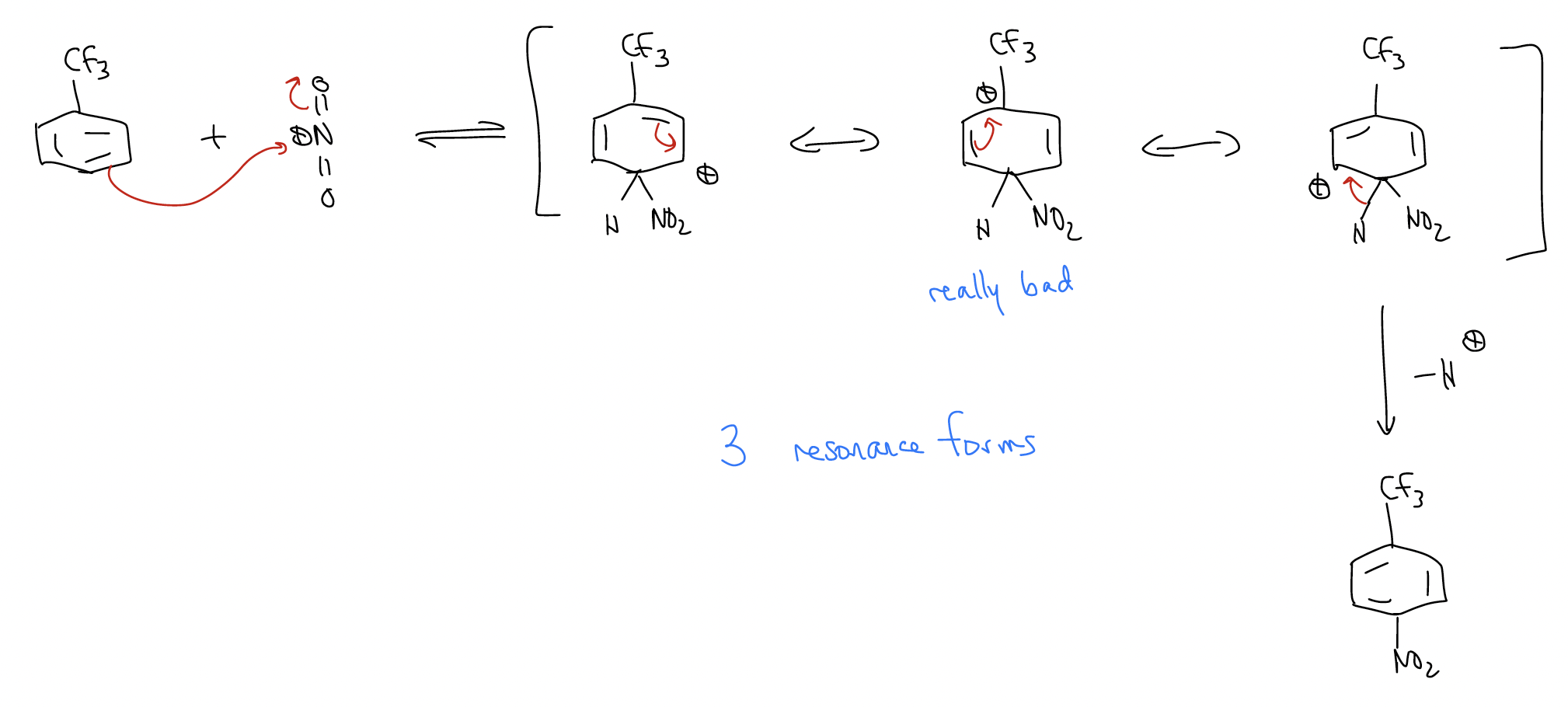
Of course, oxygen is an activator with a lone pair aligned with the \(π\) system of the benzene ring. What would happen if a deactivator was a substituent on the ring? First, the reaction would be much slower because of the deactivating group, which lowers the HOMO. Second, the deactivator would give a set of resonance forms that favor formation of the meta-substituted product. Consider the nitration of trifluorobenzene:
a) ortho-nitration
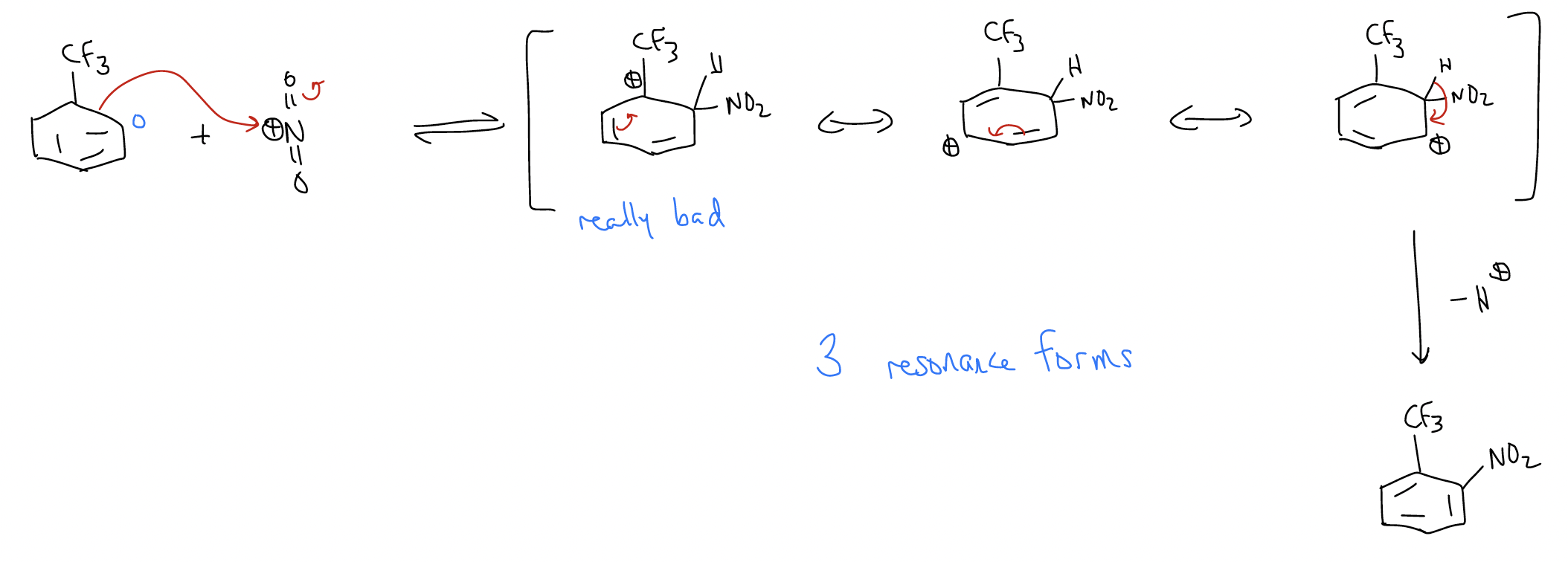
b) meta-nitration
c) para-nitration
The reaction energy diagram would look like this:
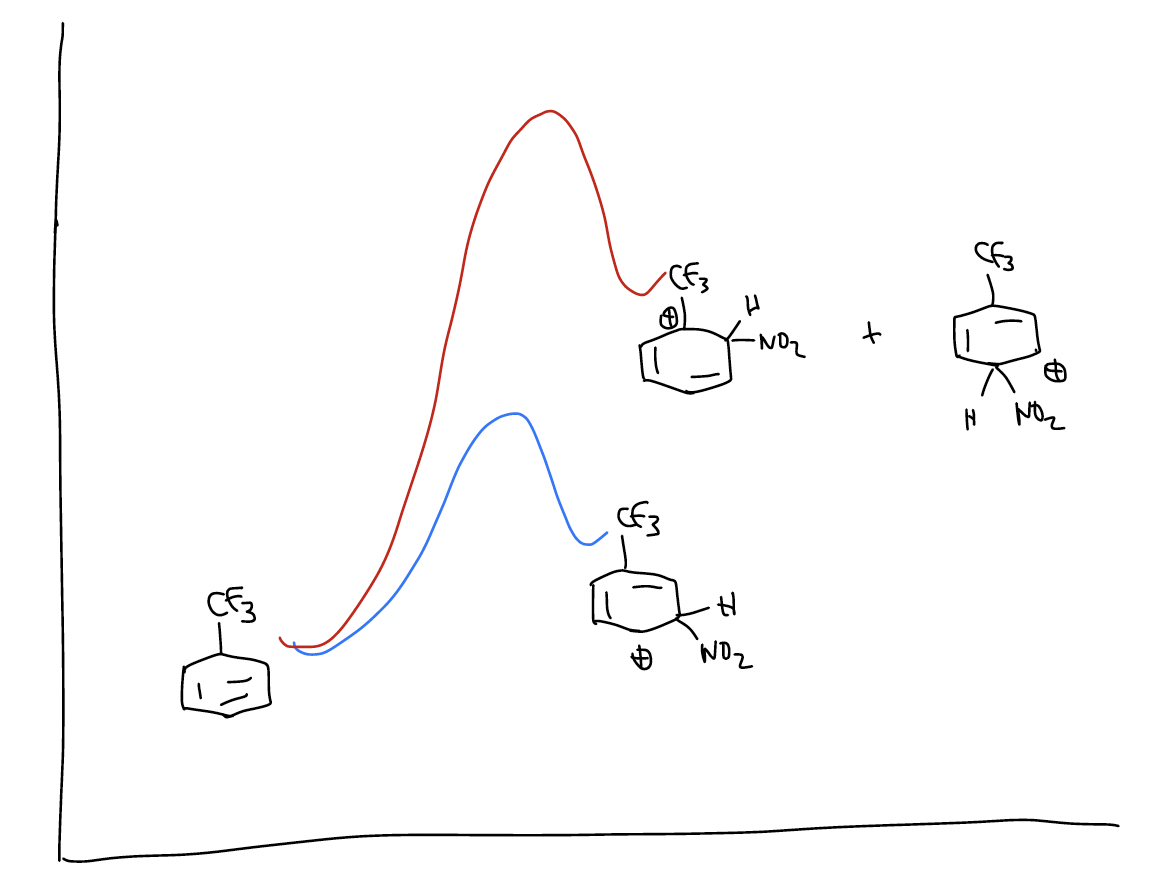
The pathway leading to the meta-product has a transition state that is lower in energy. Why? Because the trifluoromethyl group is strongly electron-withdrawing due to its inductive effect, and the arenium will sense this effect more strongly if the positive charge is on the carbon atom directly attached to it.
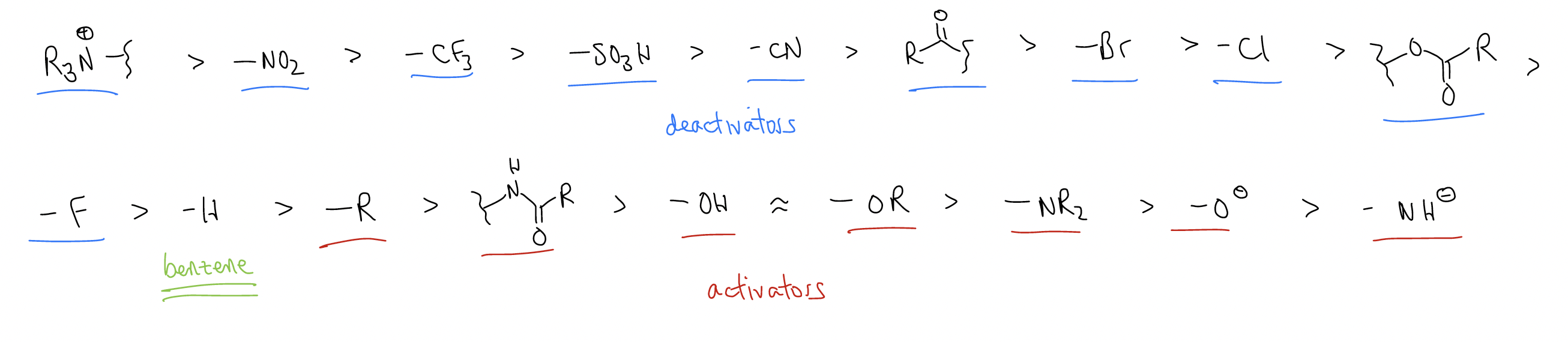
So, what does this all mean for the regioselectivity in electrophilic aromatic substitution? Can we categorize the long list of substituents not into activator/deactivator roles, but instead into ortho-/para- directors and meta-directors?
Here are some rules:
1. all activators are ortho-/para-directors
2. all substituents with lone pairs adjacent to the ring are ortho-/para-directors
3. deactivators without lone pairs adjacent to the ring are meta-directors
Can you assign the ortho-/para- or meta-directing ability in the reactivity series?