
10.7: Benzene and Aromaticity
- Page ID
- 210876
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The previous sections addressed the \(\pi\) orbitals of linear conjugated system. Here we address conjugated systems of cyclic conjugated hydrocarbons with the general formula of \(C_nH_n\) where \(n\) is the number of carbon atoms in the ring. The molecule from this important class of organic molecule that you are most familiar with is benzene (\(C_6H_6\)) with \(n=6\), although many other molecules exist like cyclobutadiene (\(C_4H_4\)) with \(n=4\) (Figure \(\PageIndex{1}\)).
The structure of benzene is an interesting historical topic. In 1865, the German chemist Friedrich August Kekulé published a paper suggesting that the structure of benzene contained a ring of six carbon atoms with alternating single and double bonds. Within this argument, two resonance structures can be formulated.
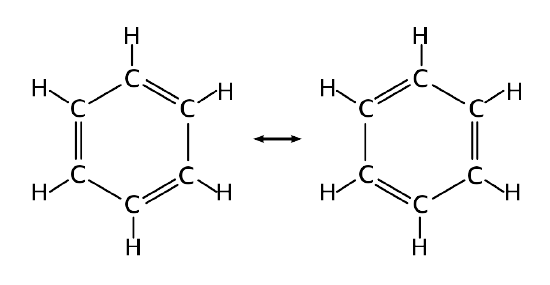
However, X-ray diffraction shows that all six carbon-carbon bonds in benzene are of the same length, at 140 pm. The C–C bond lengths are greater than a double bond (135 pm), but shorter than a typical single bond (147 pm). This means that neither structures Figure \(\PageIndex{2}\) are correct and the true 'structure' of benzene is a mixture of the two. As discussed previously, that such a valence bond perspective results in a delocalization energy within a molecular orbital approach.
Aromatic systems provide the most significant applications of Hückel theory. For benzene, we find the secular determinant
\[\left|\begin{array}{cccccc}x&1&0&0&0&1\\1&x&1&0&0&0\\0&1&x&1&0&0\\0&0&1&x&1&0\\0&0&0&1&x&1\\1&0&0&0&1&x\end{array}\right|=0\label{31}\]
with the six roots \(x=\pm2,\pm1,\pm1\). This corresponds to the following energies (ordered from most stable to least since \(\beta < 0\)):
- \(E_1 = α + 2β\)
- \(E_2 = α + β\)
- \(E_3 = α + β\)
- \(E_4 = α − β\)
- \(E_5 = α − β\)
- \(E_6 = α − 2β\)
The two pairs of \(E=\alpha\pm\beta\) energy levels are two-fold degenerate (Figure \(\PageIndex{3}\)).
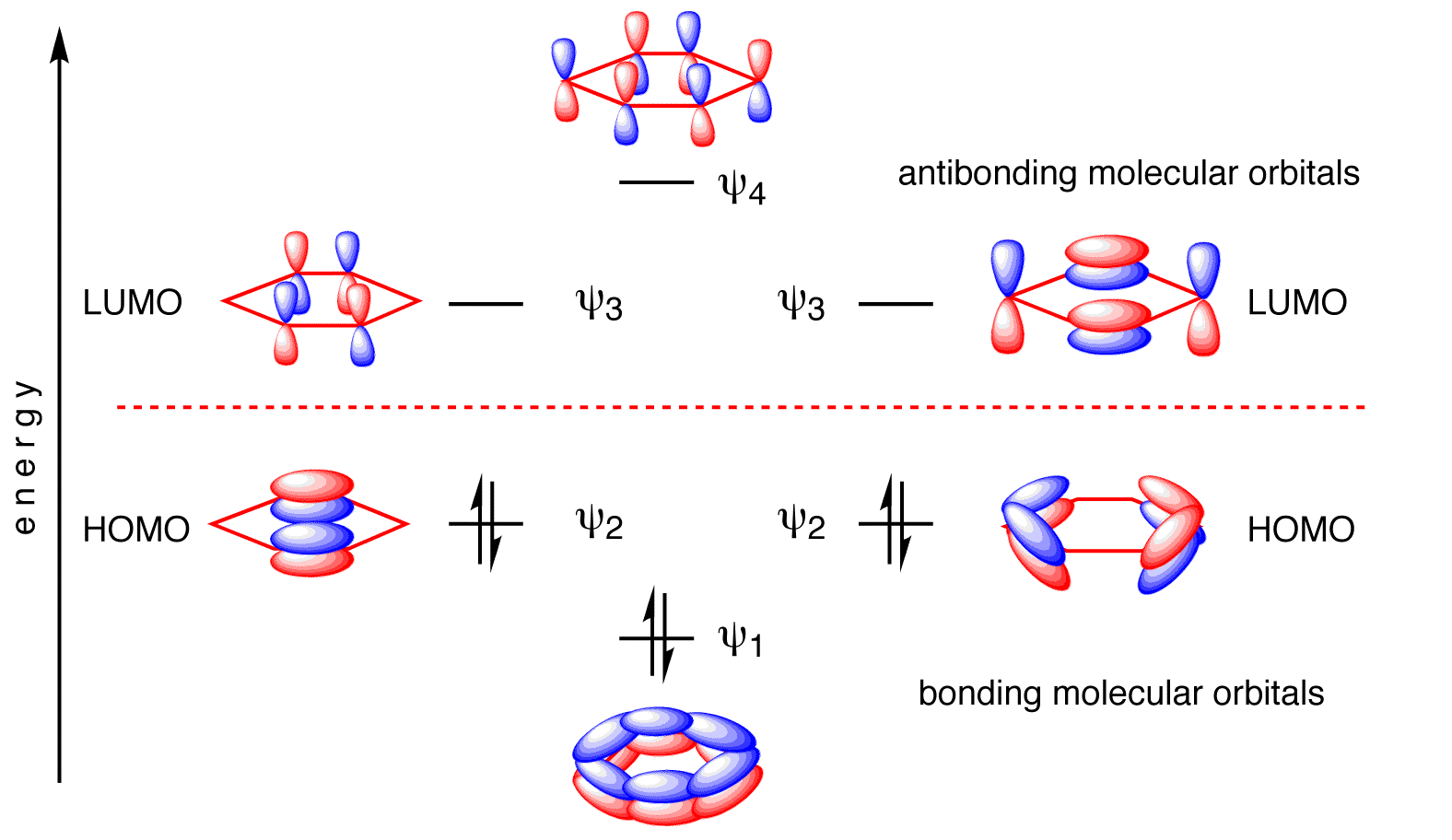
The resulting wavefunctions are below (expanded in terms of carbon \(| 2p\rangle\) atomic orbitals).
\[ \begin{align} | \psi_1 \rangle &= \dfrac{1}{\sqrt{6}} \left[ | 2p_{z1} \rangle+ | 2p_{z2} \rangle + | 2p_{z3} \rangle + | 2p_{z4} \rangle + | 2p_{z5} \rangle + | 2p_{z6} \rangle \right] \\ | \psi_2 \rangle &= \dfrac{1}{\sqrt{4}} \left[ | 2p_{z2} \rangle + | 2p_{z3} \rangle - | 2p_{z4} \rangle - | 2p_{z5} \rangle \right] \\ | \psi_3 \rangle &= \dfrac{1}{\sqrt{3}} \left[ | 2p_{z1} \rangle + \dfrac{1}{2}| 2p_{z2} \rangle - \dfrac{1}{2} | 2p_{z3} \rangle - | 2p_{z4} \rangle - \dfrac{1}{2} | 2p_{z5} \rangle + \dfrac{1}{2} | 2p_{z6} \rangle \right] \\ | \psi_4 \rangle &= \dfrac{1}{\sqrt{4}} \left[ | 2p_{z2} \rangle - | 2p_{z3} \rangle + | 2p_{z4} \rangle - | 2p_{z5} \rangle \right] \\ | \psi_5 \rangle &= \dfrac{1}{\sqrt{3}} \left[ | 2p_{z1} \rangle - \dfrac{1}{2}| 2p_{z2} \rangle - \dfrac{1}{2} | 2p_{z3} \rangle + | 2p_{z4} \rangle - \dfrac{1}{2} | 2p_{z5} \rangle - \dfrac{1}{2} | 2p_{z6} \rangle \right] \\ | \psi_6 \rangle &= \dfrac{1}{\sqrt{6}} \left[ | 2p_{z1} \rangle- | 2p_{z2} \rangle + | 2p_{z3} \rangle - | 2p_{z4} \rangle + | 2p_{z5} \rangle - | 2p_{z6} \rangle \right] \end{align}\]
Each of the carbons in benzene contributes one electron to the \(\pi\)-bonding framework (Figure \(\PageIndex{3}\)). This means that all bonding molecular orbitals are fully occupied and benzene then has an electron configuration of \(\pi_1^2 \pi_2^2 \pi_3^2 \). With the three lowest molecular orbitals occupied, the total \(\pi\)-bonding energy is
\[E_{tot} (benzene)=2(\alpha+2\beta)+4(\alpha+\beta)=6\alpha+8\beta\label{32}\]
Since the energy of a localized double bond is \( 2(\alpha+\beta)\), as determined from the analysis of ethylene, the delocalization energy of benzene is
\[ \Delta E = E_{tot} (benzene) - 3 E_{tot} (ethylene) = (6\alpha+8\beta ) - 3 \times 2(\alpha+\beta) = 2\beta\]
The experimental thermochemical value is -152 kJ mol-1.
Aromaticity
In general, cyclic polyenes are only closed shell (i.e., each electron paired up) and extra stable for with (4n+2) \(\pi\) electrons (n=0,1,2…). These special molecules have the highest delocalization energies and are said to be “aromatic”. For benzene this is \( 2\beta\) (Equation \(\ref{32}\)), which is the energy by which the delocalized \(\pi\) electrons in benzene are more stable than those in three isolated double bonds.
A stable, closed-shell conjugated cyclic structure is obtained for molecules with (4n+ 2) electrons with n=2, 6, 10, .... electrons.
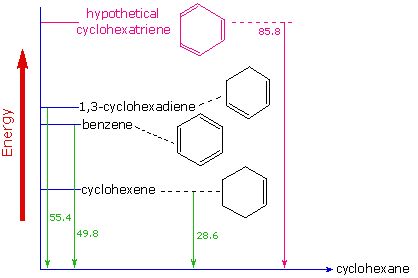
Evidence for the enhanced thermodynamic stability of benzene was obtained from measurements of the heat released when double bonds in a six-carbon ring are hydrogenated (hydrogen is added catalytically) to give cyclohexane as a common product. In the following diagram cyclohexane represents a low-energy reference point. Addition of hydrogen to cyclohexene produces cyclohexane and releases heat amounting to 11.9 kJ mol-1. If we take this value to represent the energy cost of introducing one double bond into a six-carbon ring, we would expect a cyclohexadiene to release 23.9 kJ mol-1 on complete hydrogenation, and 1,3,5-cyclohexatriene to release 35.9 kJ mol-1. These heats of hydrogenation \(\Delta H_{hyd}\) reflect the relative thermodynamic stability of the compounds (Figure \(\PageIndex{4}\)). In practice, 1,3-cyclohexadiene is slightly more stable than expected, by about 8.1 kJ mol-1, presumably due to conjugation of the double bonds. Benzene, however, is an extraordinary 15 kJ mol-1 more stable than expected. This additonal stability is a characteristic of all aromatic compounds.
Contributors
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry
- Wikipedia