
1.16: Thermochemistry
- Page ID
- 45025

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)In chemical reactions, bonds are broken and reformed, and heat may be given off or taken up in such processes. Since reactions are often carried out at constant pressure, the change in enthalpy is the appropriate measure of heat given off or absorbed.
Enthalpies of Reaction
The overall change in the enthalpy between products and reactants in a reaction is called the reaction enthalpy. The reaction enthalpy can be either positive or negative. As an example, consider the reaction:
\[CO (g) + \frac{1}{2} O_2 (g) \longrightarrow CO_2 (g) \label{17.1}\]
The change enthalpy when \(1\) mol of \(CO\) reacts completely with \(0.5\) mol of \(O_2\) at \(25^\text{o}C\) can be measured by calorimetry and is observed to be
\[\Delta H = H(\text{products}) - H(\text{reactants}) = q_P = -283.0 \: \text{kJ} \label{17.2}\]
The negative sign means that heat is given off by the system, rather than absorbed by it.
When heat is given off, the reaction is said to be exothermic. and \(\Delta H\) will be negative. If heat is absorbed, the reaction is said to be endothermic:
\[2 \: CO_2 (g) \longrightarrow 2 \: CO (g) + O_2 (g) \label{17.3}\]
the enthalpy of the reaction would be
\[\Delta H = 2 \times 283.0 \text{kJ} = 566.0 \text{kJ} \label{17.4}\]
Consider the reaction of solid phosphorus with liquid bromine:
\[2 \: P (s) + 3 \: Br_2 (l) \longrightarrow 2 \: PBr_3 (g) \: \: \: \: \: \: \: \Delta H = -243 \text{kJ} \label{17.5}\]
Suppose \(2.63\) g of phosphorus reacts with an excess of bromine. Calculate the enthalpy change.
Solution
Noting that the specified \(\Delta H\) of \(-243\) kJ refers to a reaction of \(2\) ml of solid phosphorus, we calculate the number of moles that correspond to \(2.63\) g:
\[\text{moles of P} = \frac{2.63 \text{g}}{30.97 \text{g/mol}} = 0.0849 \text{mol} \label{17.6}\]
Thus, the enthalpy change when \(0,0849\) mol of phosphorus react is
\[\Delta H = 0.0849 \text{mol} \times \left( \frac{-243 \text{kJ}}{2 \text{mol P}} \right) = -10.3 \text{kJ} \label{17.7}\]
Hess's Law
Suppose a reaction is carried out in several steps, We have already seen that the overall reaction is the algebraic sum of the individual steps. Because energy/enthalpy is an additive quantity, the overall enthalpy change for the overall reaction, obtained by adding the individual chemical equations, will be the sum of the enthalpy changes associated with each of the individual steps/chemical equations. This statement is known as Hess's Law.
Hess's Law can be used to calculate the enthalpy of reactions that cannot be carried out straightforwardly in the lab. As an example, consider the reaction of solid carbon, in the form of graphite, with oxygen gas to product carbon monoxide:
\[C (s, gr) + \frac{1}{2} O_2 \longrightarrow CO (g) \label{17.8}\]
This reaction cannot be carried out directly experimentally. Rather, a two-step process is used, in which solid carbon is first converted to carbon dioxide via the reaction
\[C (s, gr) + O_2 \longrightarrow CO_2 (g) \: \: \: \: \: \: \: \Delta H = -393.5 \text{kJ} \label{17.9}\]
whose enthalpy can be easily measured at (25^\text{o}C\). Then the decomposition of \(CO_2\) to give oxygen gas and carbon monoxide can be added to this reaction according to:
\[\begin{array}{rcll} C (s, gr) + O_2 & \longrightarrow & CO_2 (g) & \Delta H_1 = -393.5 \text{kJ} \\ CO_2 (g) & \longrightarrow & CO (g) + \frac{1}{2} O_2 & \Delta H_2 = +283.0 \text{kJ} \\ C (s, gr) + \frac{1}{2} O_2 (g) & \longrightarrow & CO (g) & \Delta H_1 + \Delta H_2 = -110.5 \text{kJ} \end{array} \label{17.10}\]
Calculate the reaction enthalpy for
\[PCl_3 (l) + Cl_2 (g) \longrightarrow PCl_5 (s) \label{17.11}\]
given that the follow reaction enthalpies are known:
\[\begin{array}{lrcll} (1) & 2 \: P (s) + 3 \: Cl_2 (g) & \longrightarrow & 2 \: PCl_3 (l) & \Delta_\text{r} H^{(1)} = -640 \text{kJ} \\ (2) & 2 \: P (s) + 5 \: Cl_2 (g) & \longrightarrow & 2 \: PCl_5 (s) & \Delta_\text{r} H^{(2)} = -887 \text{kJ} \end{array} \label{17.12}\]
In order to get these two given reactions to add to the one of interest, we first divide both reactions by \(2\):
\[\begin{array}{lrcll} (1') & P (s) + \frac{3}{2} Cl_2 (g) & \longrightarrow & PCl_3 (l) & \Delta_\text{r} H^{(1')} = -320 \text{kJ} \\ (2') & P (s) + \frac{5}{2} Cl_2 (g) & \longrightarrow & PCl_5 (s) & \Delta_\text{r} H^{(2')} = -442.5 \text{kJ} \end{array} \label{17.13}\]
Next, write reaction \((1')\) in reverse:
\[\begin{array}{lrcll} (1'') & PCl_3 (l) & \longrightarrow & P (s) + \frac{3}{2} Cl_2 (g) & \Delta_\text{r} H^{(1'')} = 320 \text{kJ} \end{array} \label{17.14}\]
Finally, when we add \((1'')\) to \((2')\), we obtain the original reaction. The overall reaction enthalpy is, therefore,
\[\Delta_\text{r} H = \Delta_\text{r} H^{(1'')} + \Delta_\text{r} H^{(2')} = 320 \text{kJ} - 443.5 \text{kJ} = -123.4 \text{kJ} \label{17.15}\]
Phase Changes
Although phase changes are not chemical reactions, a quantity of heat is still required, for example, to transform ice at \(0^\text{o}C\) to water at \(0^\text{o}C\). This quantity of heat is called the enthalpy of fusion:
\[H_2O (s) \longrightarrow H_2O (l) \: \: \: \: \: \: \: \Delta H_\text{fus} = +6.007 \text{kJ} \label{17.16}\]
Similarly, a quantity of heat is necessary to transform water at \(100^\text{o}C\) to steam at \(100^\text{o}C\). This quantity is known as the enthalpy of vaporization:
\[H_2O (l) \longrightarrow H_2O (g) \: \: \: \: \: \: \: \Delta H_\text{vap} = +40.7 \text{kJ} \label{17.17}\]
Standard-State Enthalpies
Absolute enthalpies of substances, like absolute energies, cannot be measured -- only changes in enthalpy can be measured. This, it is necessary to have a reference state, just as it is necessary to establish a reference point for computing potential energies. The reference state for reaction enthalpies is called the standard state and is defined as follows:
- For liquids and solids, the standard state is the thermodynamically stable state at a pressure of \(1\) atm and a specified temperature. The stable state could be liquid or solid.
- For gases, the standard state is the gaseous state at a pressure of \(1\) atm and a specified temperature, based on the assumption of ideal gas behavior.
- For dissolved species, the standard state is a \(1\) M solution at a pressure of \(1\) atm and specified temperature, based on the assumption of ideal solution behavior.
Given this definition of the standard state, a scale needs to be defined by arbitrarily setting the enthalpies of selected reference substances to \(0\). The choice of reference is the following: the enthalpies of chemical elements in their standard states at \(298.15\) K are defined to be \(0\). For elements that can exist in several forms, we choose that form which is most stable at \(1\) atm and \(298.15\) K. For example, oxygen can exist as \(O_2 (g)\) or \(O_3 (g)\). The former is more stable, so it is defined to have a \(0\) enthalpy. Carbon can exist as graphite, diamond, or fullerene. Graphite is the most stable of these at \(1\) atm and \(298.15\) K, sot it is assigned an enthalpy of \(0\). For any reaction, the standard state enthalpy is denoted \(\Delta H^\text{o}\).
In order to have useful enthalpy data to tabulate, one more concept is introduced. the standard enthalpy of formation of a compound is the enthalpy change for a reaction that produces \(1\) mol of the compound from its elements at \(1\) atm pressure and \(25^\text{o}C\). For example, the standard enthalpy of formation of water is the standard enthalpy of the reaction:
\[H_2 (g) + \frac{1}{2} O_2 (g) \longrightarrow H_2O (l) \: \: \: \: \: \: \: \Delta_\text{f} H^\text{o} = -285.83 \text{kJ} \label{17.18}\]
For species that dissolve into ions in solution, ions of both positive and negative charge will be produced. Clearly, we cannot measure the enthalpy of formation of ions of only one type of charge, rather, only the sum of the enthalpies can be measured. Thus, one more arbitrary assignment must be made, namely, that the standard enthalpy of formation of \(H^+ (aq)\) is \(0\)
An example of the use of tabulated standard state enthalpies, consider the following:
What is the standard state enthalpy of the reaction
\[C (s) + CO_2 (g) \longrightarrow 2 \: CO \label{17.19}\]
We need to consider this as a two-step process, wherein \(CO_2\) is broken down into its elements, then these elements recombine to form \(CO (g)\). The two steps of the reaction are
\[\begin{align} CO_2 (g) &\longrightarrow C (s) + O_2 (g) \\ 2 \: C (s) + O_2 (g) &\longrightarrow 2 \: CO (g) \end{align} \label{17.20}\]
It is easy to verify that the sum of these two reactions reproduces the desired reaction above. In Appendix D of the book, the following standard enthalpies of formation are given:
\[\begin{array}{rcll} C (s) + O_2 (g) & \longrightarrow & CO_2 (g) & \Delta_\text{f} H^\text{o} = -393.51 \text{kJ} \\ C (s) + \frac{1}{2} O_2 (g) & \longrightarrow & CO (g) & \Delta_\text{f} H^\text{o} = -110.52 \text{kJ} \end{array} \label{17.21}\]
Thus, for the reaction we are considering, the standard enthalpy of formation is the sum of the standard enthalpies of formation for the two individual steps:
\[\Delta H^\text{o} = -\Delta_\text{f} H^\text{o} (CO_2) + 2 \Delta_\text{f} H^\text{o} (CO) = 172.47 \text{kJ} \label{17.22}\]
As a second example, consider the reaction
\[C_2 H_2 (g) + \frac{5}{2} O_2 (g) \longrightarrow 2 \: CO_2 (g) + H_2 O (l) \label{17.23}\]
Calculate the reaction enthalpy at the standard state using tabulated formation enthalpies.
The three formation reactions are
\[\begin{array}{lrcll} (1) & C (s) + O_2 (g) & \longrightarrow & CO_2 (g) & \Delta_\text{f} H^\text{o} = -393.5 \text{kJ} \\ (2) & H_2 (g) + \frac{1}{2} O_2 (g) & \longrightarrow & H_2 O (l) & \Delta_\text{f} H^\text{o} = -285.8 \text{kJ} \\ (3) & 2 \: C (s) + H_2 (g) & \longrightarrow & C_2 H_2 (g) & \Delta_\text{f} H^\text{o} = 226.7 \text{kJ} \end{array} \label{17.24}\]
We just need to combine these to give the correct overall reaction. We simply need to multiply the first reaction by \((2)\) and write reaction \((3)\) in reverse. This gives
\[\begin{array}{lrcll} (1') & 2 \: C (s) + 2 \: O_2 (g) & \longrightarrow & 2 \: CO_2 (g) & \Delta_\text{f} H^\text{o} = -787 \text{kJ} \\ (2) & H_2 (g) + \frac{1}{2} O_2 (g) & \longrightarrow & H_2 O (l) & \Delta_\text{f} H^\text{o} = -258.8 \text{kJ} \\ (3') & C_2 H_2 (g) & \longrightarrow & 2 \: C (s) + H_2 (g) & \Delta_\text{f} H^\text{o} = -226.7 \text{kJ} \end{array} \label{17.25}\]
Summing these gives the correct overall reaction. Thus, the overall reaction enthalpy is
\[\Delta_\text{r} H^\text{o} = -787 \text{kJ} - 226.7 \text{kJ} - 285.8 \text{kJ} = -1299.5 \text{kJ/mol} \label{17.26}\]
As one final consideration of standard state enthalpies, we define the bond enthalpy as the enthalpy required to break \(1\) mol of a particular chemical bond in the gas phase. Since energy is required to break a chemical bond, bond enthalpies are always positive. An example of a bond enthalpy is the breaking of \(C-H\) bond in methane:
\[CH_4 (g) \longrightarrow CH_3 (g) + H (g) \: \: \: \: \: \: \: \Delta H^{o} = 438 \text{kJ} \label{17.27}\]
Although the products in this reaction are not stable, i.e., they react quickly with other molecules, it is possible to produce such reactions in the lab, and study properties of the products over the period of their brief existence. This, then, is how bond enthalpies are tabulated. Often, bond enthalpies are tabulated as averages of a particular bond breaking, e.g. a \(C-H\) bond, over many compounds. Given the relative constancy of bond enthalpies of a particular kind of bond over different molecules, such an average is a good measure of the bond enthalpy one might expect in an arbitrary specific situation.
Temperature Dependence of Reaction Enthalpies
Recall that the constant-pressure heat capacity \(C_P\) is given by
\[C_P = \left( \frac{\partial H}{\partial T} \right)_P \label{17.28}\]
For an ideal gas, \(C_P\) is independent of temperature, however, for non-ideal systems, \(C_P\) does depend on \(T\):
\[C_P(T) = \left( \frac{\partial H}{\partial T} \right)_P \label{17.29}\]
If we wish to consider the reaction enthalpy at a temperature other than that of the standard state, where enthalpies are tabulated, we can use Equation 17.29 to obtain the enthalpy at the desired temperature. Integrating both sides of Equation 17.29 from a temperature \(T_1\) to \(T_2\), we obtain
\[\begin{align} \int_{T_1}^{T_2} C_P(T) \: dT &= \int_{T_1}^{T_2} \left( \frac{\partial H}{\partial T} \right)_P \: dT \\ \int_{T_1}^{T_2} C_P(T) \: dT &= H(T_2) - H(T_1) \end{align} \label{17.30}\]
The use of Equation 17.30, of course, assumes that the \(H(T)\) is everywhere smooth and differentiable, so that it can be integrated. However, if we plot the molar enthalpy difference \(\bar{H}(T) - \bar{H} (0)\) for a real substance such as benzene, we find a curve like that shown in the figure below.
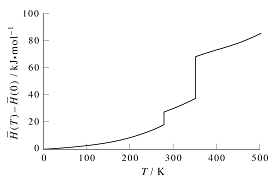
There are two temperatures at which \(H(T)\) exhibits a discontinuity. These two temperatures are the melting temperature \(T_\text{melt}\) of the solid and the vaporization temperature \(T_\text{vap}\) of the liquid. According to our discussion of phase changes, the heat needed to effect these two phase changesa re \(\Delta_\text{fus} H\) and \(\Delta_\text{vap} H\), respectively. At these temperatures, \(C_P = \partial H/\partial T\) diverges, and we cannot smoothly integrate through these divergences. In fact, divergence of the heat capacity is one of the signatures of a phase transition. As long as \(T<T_\text{melt}\) we can smoothly integrate \(C_P\) to obtain a value for \(H(T)\):
\[H(T) - H(0) = \int_0^T C_P(T') \: dT' \label{17.31}\]
However, if we are interested in a temperature \(T>T_\text{melt}\), then we must treat the discontinuity as a special point. Fortunately, we already know the enthalpy change at this point, i.e., \(\Delta_\text{fus} H\), and we can simply add it in. Thus, for \(T>T_\text{melt}\). we would calculate \(H(T)\) as follows:
\[H(T) - H(0) = \int_0^{T_\text{melt}} C_P (T') \: dT' + \Delta_\text{fus} H + \int_{T_\text{melt}}^T C_P (T') \: dT' \label{17.32}\]
The same holds for \(T>T_\text{vap}\). When we reach the discontinuity at \(T_\text{vap}\), we simply add in the enthalpy of vaporization \(\Delta_\text{vap} H\):
\[H(T) - H(0) = \int_0^{T_\text{melt}} C_P (T') \: dT' + \Delta_\text{fus} H + \int_{T_\text{melt}}^{T_\text{vap}} C_P (T') \: dT' + \Delta_\text{vap} H + \int_{T_\text{vap}}^T C_P (T') \: dT' \label{17.33}\]
Consider the reaction
\[CO_{(g)} + H_2O_{(g)} \longrightarrow H_{2(g)} + CO_{2(g)} \label{17.34}\]
For this reaction, the heat capacity has the following temperature dependence
\[C_P(T) = a + bT + cT^2 \label{17.35}\]
where \(a\), \(b\), and \(c\) are constants. Calculate the reaction enthalpy at \(800\; K\) for this reaction given the following data:
- For \(CO\): \(a = 26.86\), \(b = 6.99 \times 10^{-3}\), \(c = 8.20 \times 10^{-7}\)
- For \(H_2O\): \(a = 30.98\), \(b = 9.62 \times 10^{-3}\), \(c = 11.84 \times 10^{-7}\)\
- For \(CO_2\): \(a = 25.98\), \(b = 43.51 \times 10^{-3}\), \(c = -184.32 \times 10^{-7}\)
- For \(H_2\): \(a = 26.08\), \(b = -0.84 \times 10^{-3}\), \(c = 201.3 \times 10^{-7}\)
The units of \(a\), \(b\), and \(c\) are J \(\cdot\) mol\(^{-1} \cdot\) K\(^{-1}\), J \(\cdot\) mol\(^{-1} \cdot\) K\(^{-2}\), and J \(\cdot\) mol\(^{-1} \cdot\) K\(^{-3}\), respectively. Standard-state formation enthalpies for \(CO\), \(H_2O\), \(CO_2\), and \(H_2\) are \(-110.52\) kJ/mol, \(-241.82\) kJ/mol, \(-393.51\) kJ/mol, and \(0\) kJ/mol, respectively.
Solution
Since this is already a gas-phase reaction, we do not need to worry about melting or vaporization, so we can smoothly integrate \(C_P (T)\) from the standard state, where \(T = 298\) K to the desired \(800\; K\). Since our reference state is \(298\; K\), the integral we need is
\[\Delta_\text{f} H (800) = \Delta_\text{f} H^\text{o} + \int_{T_0}^{T_f} C_P (T) \: dT \label{17.36}\]
where \(T_0 = 298\ K\) and \(T_f = 800 \;K\). The integral is
\[\int_{T_0}^{T_f} C_P (T) \: dT = a(T_f - T_0) + \frac{1}{2} b \left( T_f^2 - T_0^2 \right) + \frac{1}{3} c \left( T_f^3 - T_0^3 \right) \label{17.37}\]
Plugging in the data, we obtain
\[\begin{align} \Delta_\text{f} H^{(800)} (CO) &= -94.981 \text{kJ/mol} \\ \Delta_\text{f} H^{(800)} (CO_2) &= -370.883 \text{kJ/mol} \\ \Delta_\text{f} H^{(800)} (H_2O) &= -223.731 \text{kJ/mol} \\ \Delta_\text{f} H^{(800)} (H_2) &= 14.668 \text{kJ/mol} \end{align} \label{17.38}\]
Therefore, the overall reaction enthalpy is
\[\begin{align} \Delta_\text{r} H^{(800)} &= \Delta_\text{f} H^{(800)} (H_2) + \Delta_\text{f} H^{(800)} (CO_2) - \Delta_\text{f} H^{(800)} (H_2O) - \Delta_\text{f} H^{(800)} (CO) \\ &= 14.668 - 370.883 + 94.981 + 223.731 = -37.503 \text{kJ/mol} \end{align} \label{17.39}\]