
16.9: Spectroscopy of Aromatic Compounds
- Page ID
- 183071
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
The chemical shifts of aromatic protons
Some protons resonate much further downfield than can be accounted for simply by the deshielding effect of nearby electronegative atoms. Vinylic protons (those directly bonded to an alkene carbon) and aromatic (benzylic) protons are dramatic examples.
We'll consider the aromatic proton first. Recall that in benzene and many other aromatic structures, a sextet of pelectrons is delocalized around the ring. When the molecule is exposed to B0, these pelectrons begin to circulate in a ring current, generating their own induced magnetic field that opposes B0. In this case, however, the induced field of the pelectrons does not shield the benzylic protons from B0 as you might expect– rather, it causes the protons to experience a stronger magnetic field in the direction of B0 – in other words, it adds to B0 rather than subtracting from it.
To understand how this happens, we need to understand the concept of diamagnetic anisotropy (anisotropy means `non-uniformity`). So far, we have been picturing magnetic fields as being oriented in a uniform direction. This is only true over a small area. If we step back and take a wider view, however, we see that the lines of force in a magnetic field are actually anisotropic. They start in the 'north' direction, then loop around like a snake biting its own tail.
If we are at point A in the figure above, we feel a magnetic field pointing in a northerly direction. If we are at point B, however, we feel a field pointing to the south.
In the induced field generated by the aromatic ring current, the benzylic protons are at the equivalent of ‘point B’ – this means that the induced current in this region of space is oriented in the same direction as B0.
In total, the benzylic protons are subjected to three magnetic fields: the applied field (B0) and the induced field from the pelectrons pointing in one direction, and the induced field of the non-aromatic electrons pointing in the opposite (shielding) direction. The end result is that benzylic protons, due to the anisotropy of the induced field generated by the ring current, appear to be highly deshielded. Their chemical shift is far downfield, in the 6.5-8 ppm region.
Characteristic NMR Absorption of Benzene Derivatives
Hydrogens directly attached to an arene ring show up about 7-9 PPM in the NMR. This is called the aromatic region. Hydrogen environments directly bonded to an arene ring show up about 2.5 PPM.

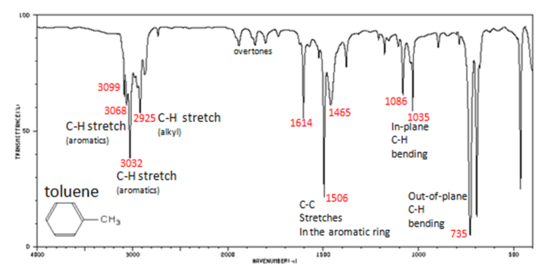
Charateristic IR Absorption of Benzene Derivatives
Arenes have absorption bands in the 650-900 cm−1 region due to bending of the C–H bond out of the plane of the ring. The exact placement of these absorptions can indicate the pattern of substitution on a benzene ring. However, this is beyond the scope of introductory organic chemistry. Arenes also possess a characteristic absorption at about 3030-3100 cm−1 as a result of the aromatic C–H stretch. It is somewhat higher than the alkyl C–H stretch (2850–2960 cm−1), but falls in the same region as olefinic compounds. Two bands (1500 and 1660 cm−1) caused by C=C in plane vibrations are the most useful for characterization as they are intense and are likely observed.
In aromatic compounds, each band in the spectrum can be assigned:
- C–H stretch from 3100-3000 cm-1
- overtones, weak, from 2000-1665 cm-1
- C–C stretch (in-ring) from 1600-1585 cm-1
- C–C stretch (in-ring) from 1500-1400 cm-1
- C–H "oop" from 900-675 cm-1
Note that this is at slightly higher frequency than is the –C–H stretch in alkanes. This is a very useful tool for interpreting IR spectra. Only alkenes and aromatics show a C–H stretch slightly higher than 3000 cm-1.
Exercise
10. A straw-colored oily liquid with a bitter almond odor and burning nut-like taste was isolated from the exudate of the castor sacs of mature North American beavers. The oily is slightly soluble in water with a boiling point of 197oC. Elemental analysis results are as follows: 68.84% C, 4.96% H, and 26.20% O. Name, draw the bond-line structure, and correlate the structure with the IR, 1H NMR, and 13C NMR spectral data below.
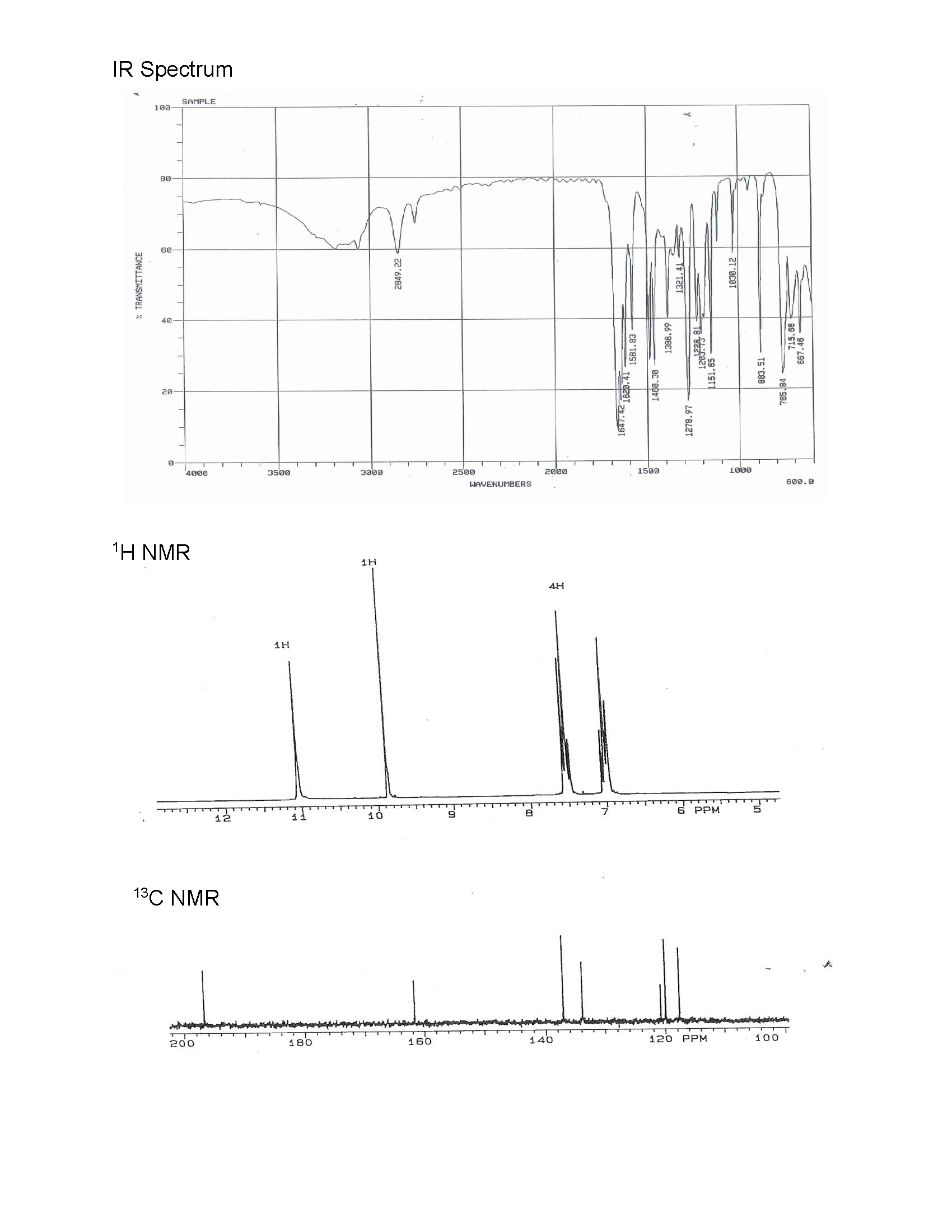
- Answer
-
10.

Contributors and Attributions
Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
Prof. Steven Farmer (Sonoma State University)
Organic Chemistry With a Biological Emphasis by Tim Soderberg (University of Minnesota, Morris)