
6.15: Characteristics of the E1 Reaction
- Page ID
- 182914
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Learning Objective
- determine the rate law & predict the mechanism based on its rate equation or reaction data for E1 reactions
- predict the products and specify the reagents for E1 reactions with stereochemistry
- propose mechanisms for E1 reactions
- draw and interpret Reaction Energy Diagrams for E1 reactions
General Reaction
Unimolecular Elimination (E1) is a reaction in which loss of the leaving group followed by removal of he beta-hydrogen results in the formation of a double bond.
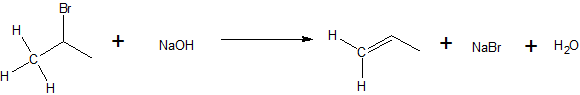
It is similar to a unimolecular nucleophilic substitution reaction (SN1) in various ways. One being the formation of a carbocation intermediate as the rate determining (slow) step, hence the name unimolecular. . Alkyl halides that can ionize to form stable carbocations are more reactive via the E1 mechanism. Because carbocation stability is the primary energetic consideration, stabilization of the carbocation via solvation is also an important consideration. Because carbocations are highly reactive, the strength of the base is not important and weak bases can be used. Since SN1 and E1 reactions behave similarly, they often compete against each other. Many times, both these reactions will occur simultaneously to form different products from a single reaction. However, one can be favored over another through thermodynamic control. Heating the reaction favors elimination over substitution.
In order of decreasing importance, the factors impacting E1 reaction pathways are
1) structure of the alkyl halide
2) stability of the carbocation
3) type of solvent
4) strength of the base.
Mechanism for Alkyl Halides
As can be seen in the E1 mechanism below, the preliminary step is the leaving group (LG) leaving on its own. Because it takes the electrons in the bond along with it, the carbon that was attached to it loses its electron, making it a carbocation. Once it becomes a carbocation, a Lewis Base (B-) deprotonates the intermediate carbocation at the beta position, which then donates its electrons to the neighboring C-C bond to form a double bond. Unlike E2 reactions, which require the proton to be anti to the leaving group, E1 reactions only require a neighboring hydrogen. This is due to the fact that the leaving group has already left the molecule. The final product is an alkene along with the HB byproduct and leaving group salt.
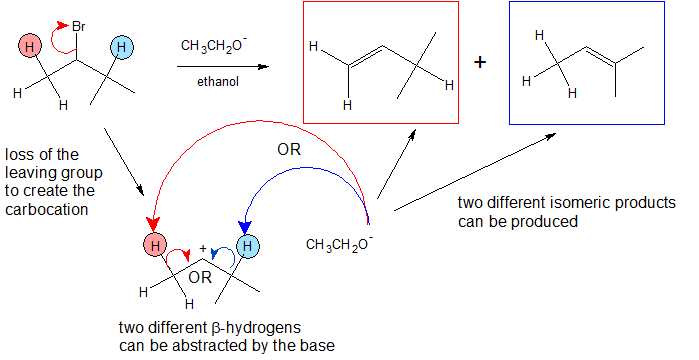
Once again, we see the two steps of the E1 mechanism.
- A base deprotonates a beta carbon to form a pi bond.
In this case we see a mixture of products rather than one discrete one. This is the case because the carbocation has two nearby carbons that are capable of being deprotonated, but that only one forms a major product (more stable).
Reactivity
Due to the fact that E1 reactions create a carbocation intermediate, rules present in \(S_N1\) reactions still apply.
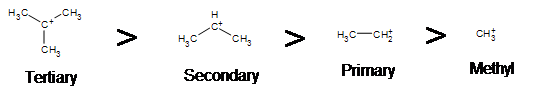
As expected, tertiary carbocations are favored over secondary, primary and methyl’s. This is due to the phenomena of hyperconjugation, which essentially allows a nearby C-C or C-H bond to interact with the p orbital of the carbon to bring the electrons down to a lower energy state. Thus, this has a stabilizing effect on the molecule as a whole. In general, primary and methyl carbocations do not proceed through the E1 pathway for this reason, unless there is a means of carbocation rearrangement to move the positive charge to a nearby carbon. Secondary and Tertiary carbons form more stable carbocations, thus this formation occurs quite rapidly.
Secondary carbocations can be subject to the E2 reaction pathway, but this generally occurs in the presence of a good / strong base. Adding a weak base to the reaction disfavors E2, essentially pushing towards the E1 pathway. In many instances, solvolysis occurs rather than using a base to deprotonate. This means heat is added to the solution, and the solvent itself deprotonates a hydrogen. The medium can effect the pathway of the reaction as well. Polar protic solvents may be used to hinder nucleophiles, thus disfavoring E2 / Sn2 from occurring.
Regiochemistry & Stereochemistry of the E1 Reaction
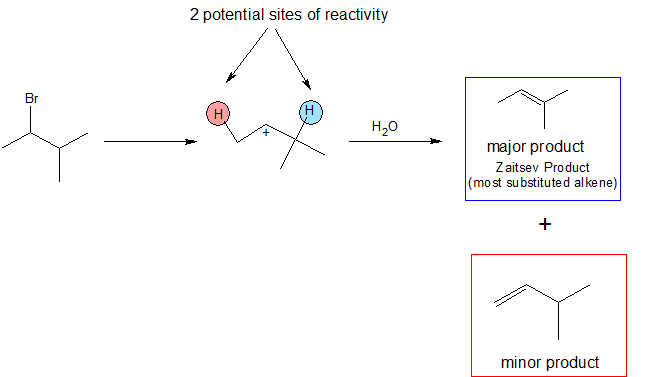
The E1 reaction is regiospecific because it follows Zaitsev's rule that states the more substituted alkene is the major product. This infers that the hydrogen on the most substituted carbon is the most probable to be deprotonated, thus allowing for the most substituted alkene to be formed.
Unlike E2 reactions, the E1 reaction is not stereospecific. Thus, a hydrogen is not required to be anti-coplanar to the leaving group because the leaving group is gone. In the mechanism below, we can see two possible pathways for the reaction. Either one leads to a plausible resultant product, however, only one forms a major product. As stated by Zaitsev's rule, deprotonation of the most substituted carbon results in the most substituted alkene. This then becomes the most stable product due to hyperconjugation, and is also more common than the minor product.
Exercises
- Which of these steps is the rate determining step (A or B)?
What is the major product formed (C or D)?
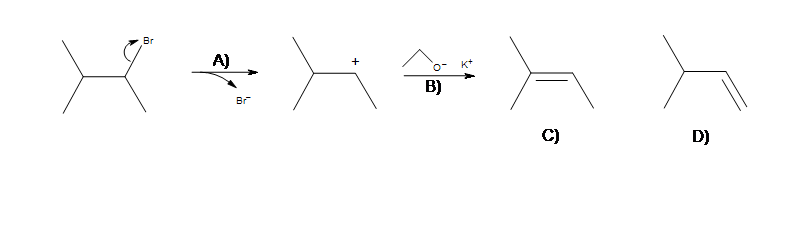
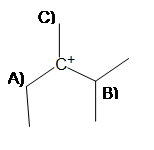
2. In order to produce the most stable alkene product, from which carbon should the base deprotonate (A, B, or C)?
If the carbocation were to rearrange, on which carbon would the positive charge go onto without sacrificing stability (A, B, or C)?
3) Predict the major product of the following reaction.
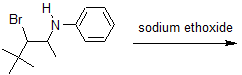
4) (True or False) – There is no way of controlling the product ratio of E1 / Sn1 reactions.
5) Explain why the presence of a weak base / nucleophile favors E1 reactions over E2.
- Answer
-
1. A , C
2. B, B
3.
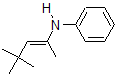
4. False - They can be thermodynamically controlled to favor a certain product over another.
5. By definition, an E1 reaction is a Unimolecular Elimination reaction. This means the only rate determining step is that of the dissociation of the leaving group to form a carbocation. Since E2 is bimolecular and the nucleophilic attack is part of the rate determining step, a weak base/nucleophile disfavors it and ultimately allows E1 to dominate. (Don't forget about SN1 which still pertains to this reaction simaltaneously).
Outside Links
- E1 reaction background: http://en.Wikipedia.org/wiki/E1_elimination
Outside Sources
- McMurry, J., Simanek, E. Fundamentals of Organic Chemistry, 6th edition. Cengage Learning, 2007.
Contributors
- Satish Balasubramanian