
10.5: Electrophilic Substitution
- Page ID
- 242298

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Until now, have already been introduced to electrophilic addition and electrophilic isomerization - now, let's move to the third variation on the electrophilic theme, that of electrophilic substitution. In an electrophilic substitution reaction, a pair of \(\pi\)-bonded electrons first attacks an electrophile - usually a carbocation species - and a proton is then abstracted from an adjacent carbon to reestablish the double bond, either in the original position or with isomerization.
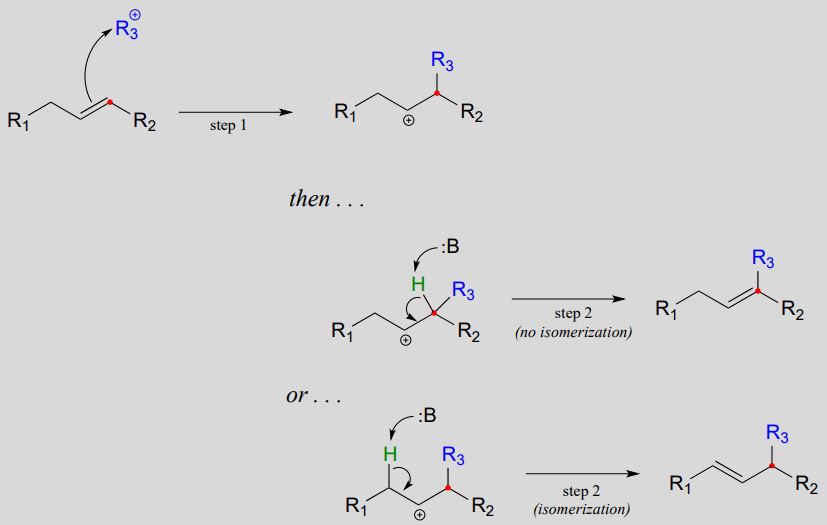
Electrophilic substitution mechanism:
Electrophilic substitution reactions in isoprenoid biosynthesis
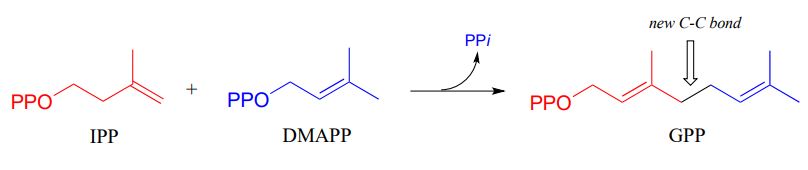
Electrophilic substitution steps are very important in the biosynthetic pathways if isoprenoid compounds. In an early, chain-elongating reaction (EC 2.5.1.1) of the pathways of many isoprenoids, building blocks IPP and DMAPP combine to form a 10-carbon isoprenoid product called geranyl diphosphate (GPP):
In a preliminary step (step a below), the diphosphate group on DMAPP departs to form an allylic carbocation.
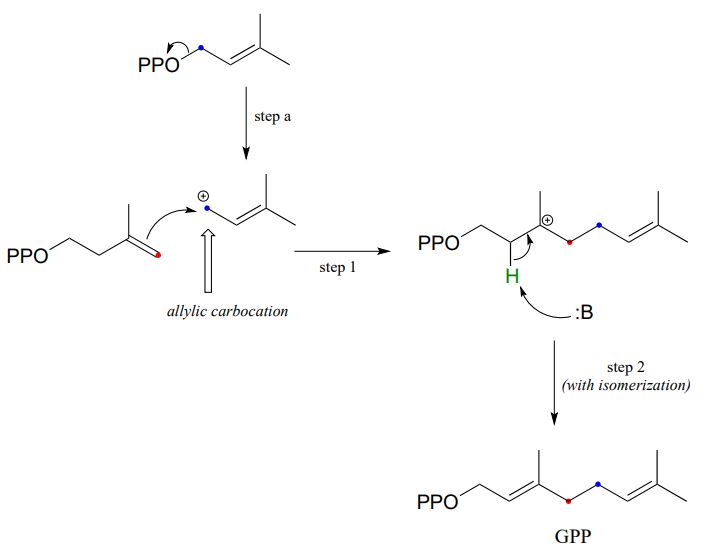
In step 1, the \(\pi \) electrons in IPP then attack the electrophilic carbocation from step a, resulting in a new carbon-carbon bond and a tertiary carbocation intermediate. Proton abstraction (step 2) leads to re-establishment of a double bond one carbon over from where it started out in IPP.
DMAPP is much more prone to spontaneous hydrolysis than IPP when they are dissolved in water. Explain why.
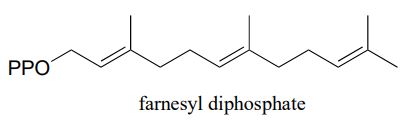
Farnesyl diphosphate (FPP) is synthesized by adding another five-carbon building block to geranyl diphosphate. What is this building block - IPP or DMAPP? Draw a mechanism for the formation of FPP.
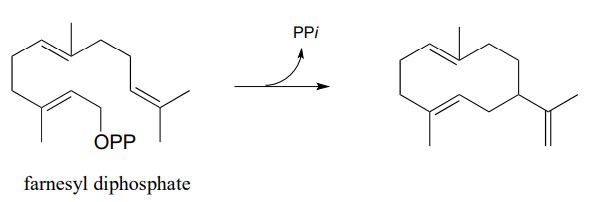
Propose a likely mechanism for the following transformation, which is the first stage in a somewhat complex reaction in the synthesis of an isoprenoid compound in plants. (Science 1997, 277, 1815)
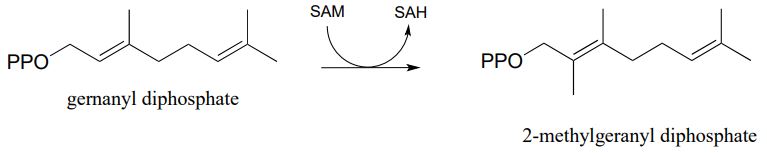
The electrophilic carbon in an electrophilic substitution reaction is often a carbocation, but it can also be the methyl group on S-adenosylmethionine (SAM - see section 8.8A). Propose a likely mechanism for this methylation reaction. (Biochemistry 2012, 51, 3003)
Electrophilic aromatic substitution
Until now, we have been focusing mostly on electrophilic reactions of alkenes. Recall from section 2.2 that \(\pi \) bonds in aromatic rings are substantially less reactive than those in alkenes. Aromatic systems, however, do in fact undergo electrophilic substitution reactions given a powerful electrophile such as a carbocation, and if the carbocation intermediate that forms can be sufficiently stabilized.
Electrophilic aromatic substitution (Friedel-Crafts alkylation) mechanism
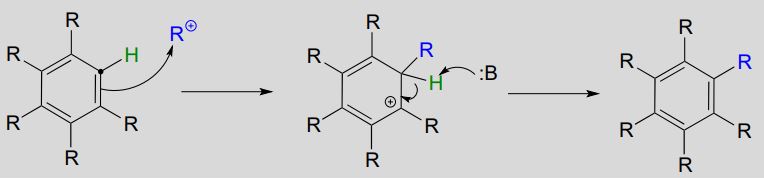
Organic chemists often refer to electrophilic aromatic substitution reactions with carbocation electrophiles as Friedel-Crafts alkylation reactions.
Aromatic rings generally do not undergo electrophilic addition reactions. Why not?
The Friedel-Crafts reaction below is part of the biosynthesis of vitamin K and related biomolecules.
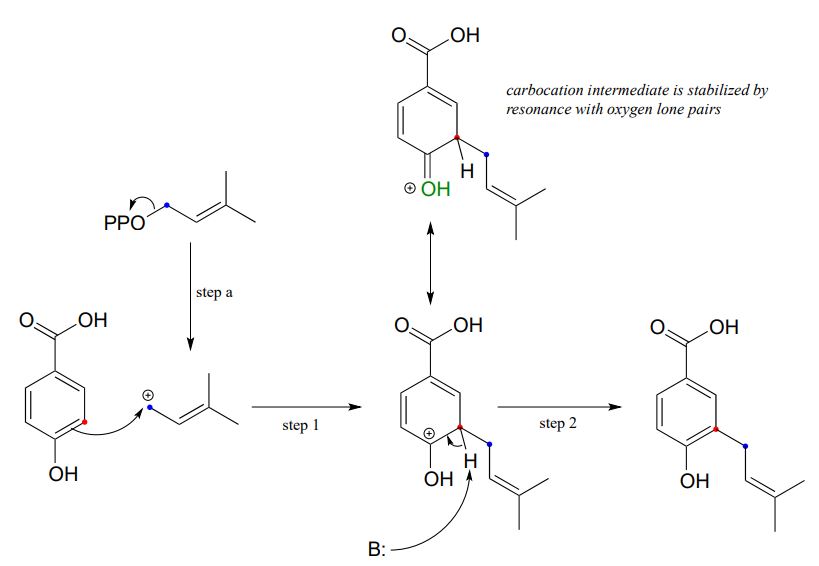
Loss of diphosphate creates a powerful carbocation electrophile (step a) which attracts the \(\pi \) electrons of the aromatic ring to form a carbocation intermediate with a new carbon-carbon bond (step 1). Substitution is completed by proton abstraction (step 2) which re-establishes the aromatic sextet.
An important point must be made here: because aromatic \(\pi \) bonds are substantially less reactive than alkene p bonds, the electrophilic must be VERY electrophilic - usually a carbocation. In addition, the carbocation intermediate that results from attack by aromatic \(\pi \) electrons is generally stabilized by resonance with lone pair electrons on a nearby oxygen or nitrogen (look at the resonance forms of the positively-charged intermediate that forms as the result of step 1 in the above figure).
Remember that stabilizing the intermediate formed in a rate-limiting step has the effect of lowering the activation energy for the step, and thus accelerating the reaction.
Organic chemists use the term ring activation to refer to the rate-accelerating effect of electron-donating heteroatoms in electrophilic aromatic substitution reactions. Aromatic rings lacking any activating oxygen or nitrogen atoms are less reactive towards electrophilic substitution.
An example of the ring-activating effect of the nitrogen atom on an aromatic ring can be found in the following Friedel-Crafts reaction (EC 2.5.1.34), which should be familiar from the introduction to this chapter:
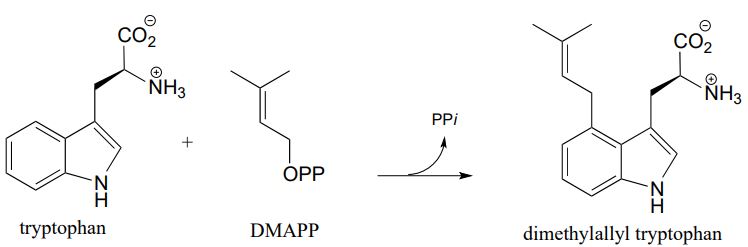
Recall that this is a key early step in the biosynthetic pathway for the ergot alkaloids which are hypothesized to have been the root cause of the 'bewitchment' of several young girls in 17th century Salem, Massachusetts. (J. Am. Chem. Soc. 1992,114, 7354).
Draw a likely mechanism for the biosynthesis of dimethylallyl tryptophan, including a resonance structure showing how the carbocation intermediate in the rate determining step is stabilized by lone pair electrons on the ring nitrogen (in other words, show how the nitrogen serves to activate the ring).
Friedel-Crafts reactions, in addition to being important biochemical transformations, are commonly carried out in the laboratory. It is instructive to consider a few examples to see how the same principles of structure and reactivity apply to both biochemical and laboratory reactions.
Below is an example of a laboratory Friedel-Crafts alkylation reaction:
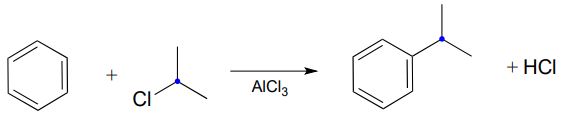
Recall that a powerful electrophile - such as a carbocation - is required for an electrophilic aromatic substitution to occur. The 2-chloropropane reactant is electrophilic, but not electrophilic enough to react with benzene. Here's where the aluminum trichloride catalyst comes in: it reacts as a Lewis acid with the alkyl chloride to generate a secondary carbocation:
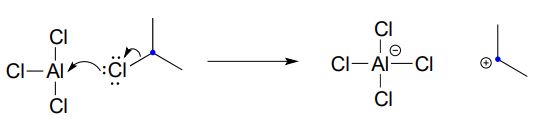
The carbocation thus generated is sufficiently electrophilic to react with the aromatic \(\pi \) electrons, in a manner that should be familiar from the biochemical examples discussed above:
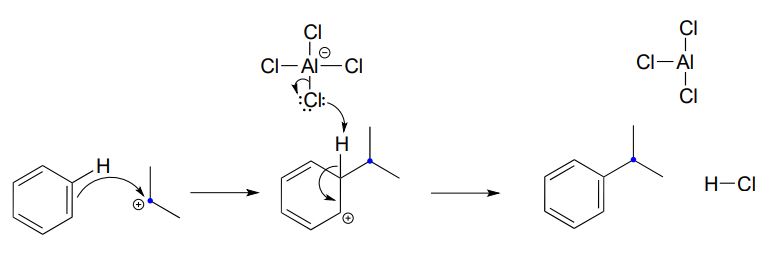
You may have noticed, however, that one element from the biochemical Friedel-Crafts reactions is missing here: there is no activating group to stabilize the ring carbocation intermediate. Indeed, the presence of an activating group - for example, the oxygen atom of a methoxy substituent - greatly increases the rate of a Friedel-Crafts alkylation.
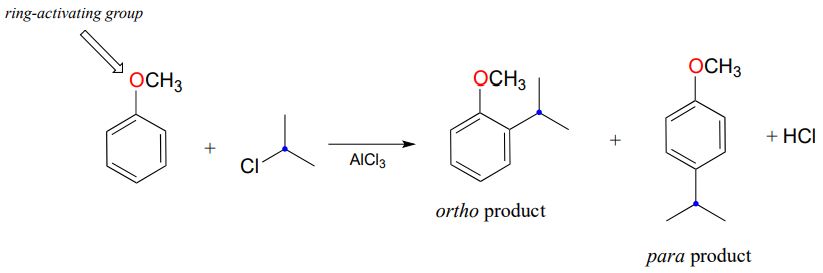
Note in the example shown above that two products are formed: one is an ortho-disubstituted benzene and one is para-disubstituted. Note also that no meta-disubstituted product is formed. This phenomenon is referred to as the ortho-para directing effect, and you are led towards an explanation in the exercise below.
- Draw the lowest-energy resonance contributors of the carbocation intermediates leading to formation of the ortho and para products in the reaction above. Use resonance structures to illustrate how the methoxy substituent is a ring-activating group.
- Draw the hypothetical carbocation intermediate in a reaction leading to formation of a meta-disubstituted product. Is this carbocation stabilized by the methoxy oxygen? Can you see why no meta product forms?
- a) Just as there are ring-activating groups in electrophilic aromatic substitutions, there are also ring-deactivating groups. For each of the substituted benzene reactants below, draw the carbocation intermediate leading to the ortho substitution product and decide whether the substituent is ring-activating or ring-deactivating in a Friedel-Crafts reaction with 2-chloropropane and AlCl3 (in other words, which compounds would react faster than benzene, and which would react slower?) Explain how the ring-deactivating effect works.
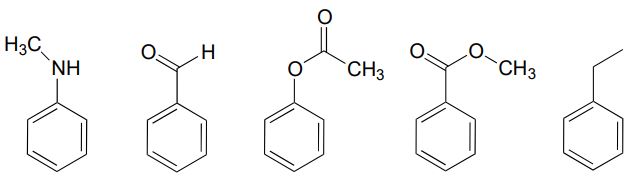
- (challenging!) Ring-deactivating substituents are usually also meta-directing. Use one of your carbocation intermediate drawings from part (a) of this exercise, and the concept od resonance, to explain this observation.
- (answer part (b) first) Look again at the vitamin K biosynthesis reaction, and discuss the ring activating/directing effects of the two substituents on the substrate.