
7.8: Biological Nucleophilic Substitution Reactions
- Page ID
- 242265

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The nucleophilic substitution reactions we have seen so far have all been laboratory reactions, rather than biochemical ones. Now, finally, let's take a look at a few examples of nucleophilic substitutions in a biological context. All of the principles we have learned so far still apply to these biochemical reactions, but in addition we need to consider the roles of the enzyme catalysts.
This is the first time that we will be seeing 'real' biological organic reaction mechanisms. Do not be intimidated by the size and complexity of the reacting biomolecules - they are just organic molecules, with the same bonding patterns and functional groups that you are already familiar with. Focus on the reacting parts of the molecule: what is the nucleophile? The electrophile? The leaving group? In most biological organic reactions, the main bulk of the biomolecule is just 'going along for the ride', and can often be abbreviated with an 'R group' (section 1.2) to simplify the picture.
A biochemical \(S_N2\) reaction
One very important class of nucleophilic substitution reactions in biochemistry are the \(S_N2\) reactions catalyzed by \(S\)-adenosyl methionine (SAM) – dependent methyltransferase enzymes. SAM is a coenzyme (section 6.3) that plays the role of methyl group donor: you can think of SAM in this context as being simply a methyl carbon electrophile attached to a sulfide leaving group.
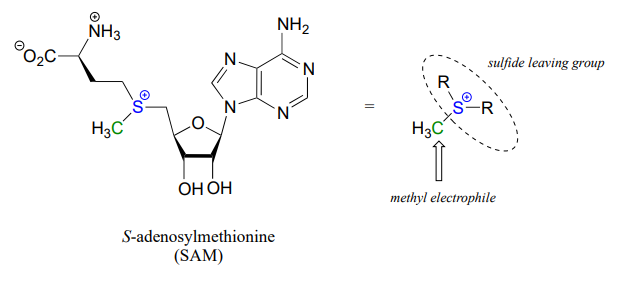
There are many variations of SAM-dependent methylation reactions in nature. In the introduction to this chapter, we were introduced to a reaction occurring in bacterial DNA in which a methyl carbon is transferred from SAM to a nitrogen atom on adenine (this type of reaction is often referred to as \(N\)-methylation).
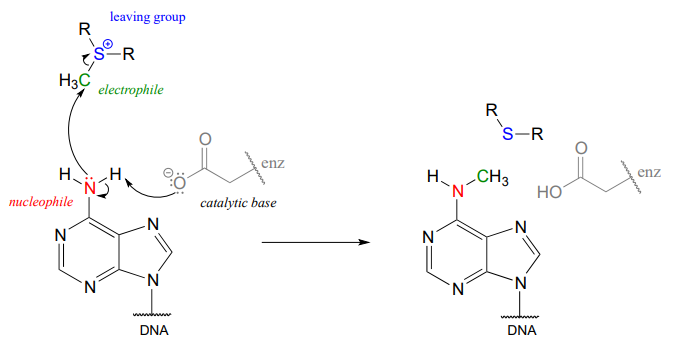
In the figure above, we are showing how an aspartate residue in the active site of the enzyme acts as a catalytic base: transfer of a proton from substrate to the aspartate side chain begins to enhance the nucleophilicity of the amine nitrogen as it approaches the electrophilic methyl carbon of SAM, and formation of the new \(N-C\) bond and cleavage of the \(C-S\) bond begins. These four bond-rearranging events probably take place in concerted fashion. A likely transition state is approximated below:
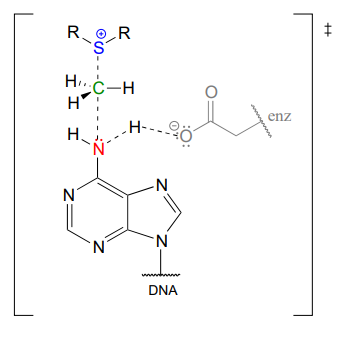
Of course, there are many other noncovalent interactions between active site enzyme residues and the substrate (the adenine base) and cofactor (SAM), but in the interest of clarity these are not shown. These interactions, many of which are hydrogen-bonds, help to position the adenine base and SAM in just the right relative orientation inside the active site for the nucleophilic attack to take place. (If you have access to American Chemical Society journals, a paper about an enzyme catalyzing a similar N-methylation reaction contains some detailed figures showing hydrogen-bond and charge-dipole interactions between the enzyme active site and the two substrates: see Biochemistry 2003, 42, 8394, figure 4).
The electrophile is a methyl carbon, so there is little steric hindrance to slow down the nucleophilic attack. The carbon is electrophilic (electron-poor) because it is bonded to a positively-charged sulfur, which is a powerful electron withdrawing group. The positive charge on the sulfur also makes it an excellent leaving group, because as it leaves, it becomes a neutral and very stable sulfide. All in all, we have a good nucleophile (enhanced by the catalytic base), an unhindered electrophile, and an excellent leaving group. We can confidently predict that this reaction is \(S_N2\). An \(S_N1\) mechanism is extremely unlikely: a methyl cation is very unstable and thus is not a reasonable intermediate to propose.
Notice something else about the SAM methylation mechanism illustrated in the previous figure. It is termolecular: there are three players acting in concert: the catalytic base, the nucleophile, and the electrophile. This is possible because the all three players are bound in a very specific geometry in the active site of the enzyme. In a reaction that takes place free in solution, rather than in an active site, the likelihood of three separate molecules colliding all at once, with just the right geometry for a reaction to take place, is very, very low. You should notice going forward that when we illustrate the mechanism of a reaction that takes place free in solution, we will only see bimolecular steps - two molecules colliding. Almost all of the biochemical reactions we see in this book will be enzyme-catalyzed - and termolecular steps will be common - while almost all of the laboratory reactions we see will take place free in solution, so we will only see unimolecular and bimolecular steps. (Synthetic chemists often employ non-biological catalysts that mimic enzyme active sites, but these examples are well beyond the scope of our discussion).
Think back to the acid-base chapter: the \(pK_a\) of a protonated ether is approximately zero, indicating that an ether is a very weak base. Considering periodic trends in acidity and basicity, what can you say about the relative basicity of a sulfide?
Another SAM-dependent methylation reaction is catalyzed by an enzyme called catechol-\(O\)-methyltransferase. The substrate here is epinephrine, also known as adrenaline, and the reaction is part of the pathway by which adrenaline is degraded in the body.
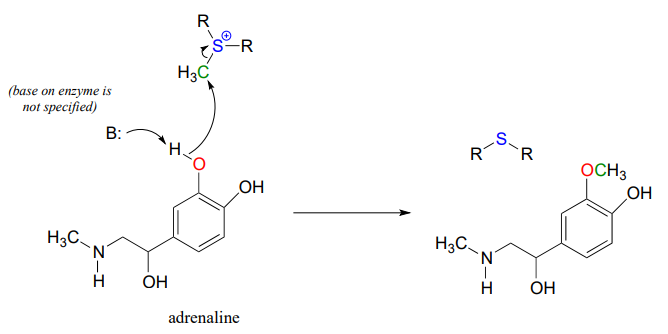
Notice that in this example, the attacking nucleophile is a phenol oxygen rather than a nitrogen (that’s why the enzyme is called an \(O\)-methyltransferase). In many cases when drawing biochemical reaction mechanisms, we use the abbreviations B: for a catalytic base and \(H-A\) for a catalytic acid, in order to keep the drawings from getting too 'busy' (it's also possible that the identity of the acidic or basic group may not be known).
SAM is formed by a nucleophilic substitution reaction between methionine and adenosine triphosphate (ATP). Draw a mechanism for this reaction, and explain why you chose either an \(S_N1\) or and \(S_N2\) pathway.
A biochemical \(S_N1\) reaction
As we will see in chapter 10, enzyme-catalyzed \(S_N1\) reactions play a critical role in carbohydrate and DNA/RNA nucleotide metabolism. The reaction below is part of nucleotide biosynthesis:
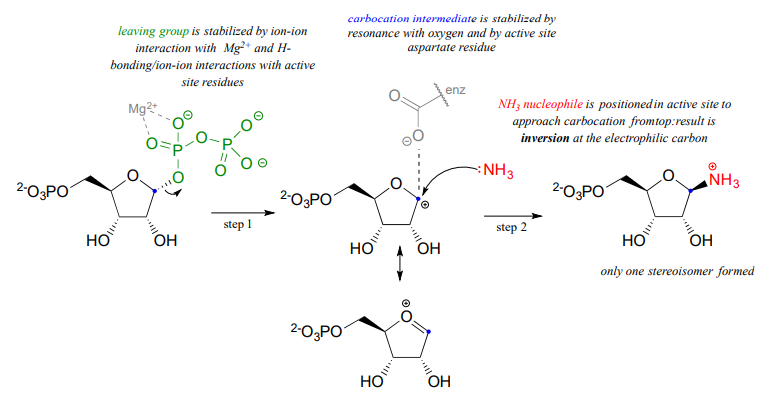
Notice a few things here: first, the diphosphate leaving group is stabilized by interactions with \(Mg^{+2}\) ion bound in the active site and also by hydrogen-bonding with active site amino acid residues (not shown). The carbocation intermediate is stabilized by resonance with the lone pairs on the oxygen (see section 8.5), and also by an active site aspartate side chain. The ammonia nucleophile is positioned in the active site so that it approaches from the 'top' side of the planar carbocation intermediate, and the substitution results in inversion of configuration. Remember: \(S_N1\) reactions which occur free in solution tend to result in a mixture of stereoisomers, but enzyme-catalyzed reactions - including enzymatic \(S_N1\) reactions such as this one - are generally stereo- and regio-specific, meaning that they almost always result in a single isomeric product, not a mixture of products.
Recall the statement from section 8.4 that poor leaving groups often need to be converted into good leaving groups. Backing up one metabolic step from the reaction depicted above, we see that a poor (hydroxide) leaving group on ribose-5-phosphate is first converted to a good (diphosphate) leaving group, which can stabilized through interactions with the active site of the enzyme catalyzing the \(S_N1\) reaction.
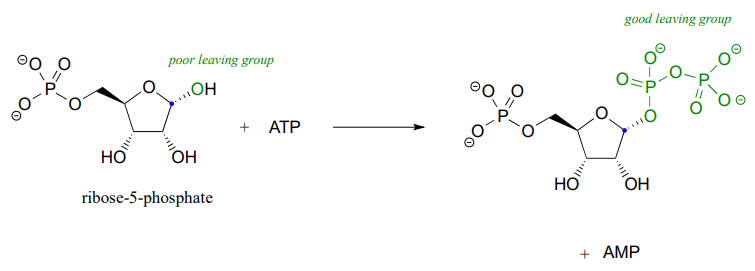
This preliminary phosphorylation step, which requires ATP (adenosine triphosphate) as the donor of the diphosphate group, is a reaction that we will study in much more detail in chapter 9.
A biochemical \(S_N1/S_N2\) hybrid reaction
The cysteine residues of certain proteins are modified by addition of a 15-carbon isoprene chain (section 1.3) to the side chain thiol group.
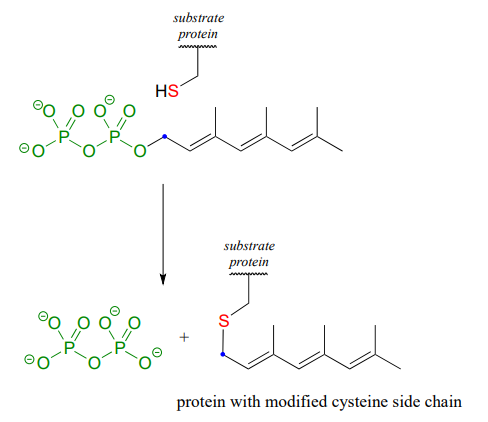
The mechanistic details of this reaction are of particular interest to biomedical scientists. The proteins that are substrates for this type of modification are involved in cell signaling processes, and they are not able to carry out their biological functions unless they are anchored to a cell’s lipid membrane. The hydrocarbon group that becomes attached to a cysteine residue in this reaction serves as the anchor.
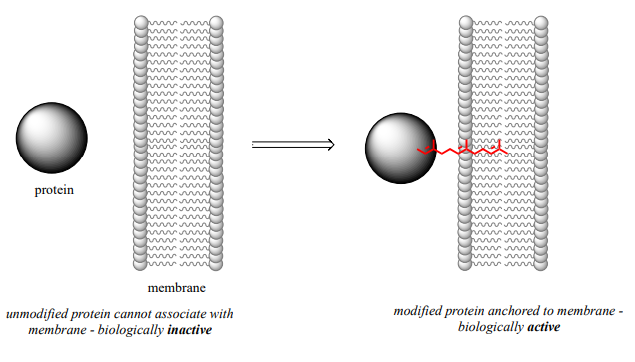
Some of these proteins have been implicated in tumor formation. Scientists hope that if they can find a way to shut down the cysteine modification reaction, the tumor-causing proteins will not be able to anchor to cell membranes and thus will remain inactive. The search is on for an effective inhibitor of this enzyme to serve as a potential anti-tumor drug.
How does the enzyme lower the energy barrier for this reaction? Experimental evidence indicates that when a substrate protein is bound to the active site of the enzyme, the cysteine thiol associates with a zinc ion bound in the active site. As we learned in section 7.8, this association will lower the pKa of the thiol to the point where it loses a proton and exists as a thiolate anion in the active site - a thiolate is a very potent nucleophile! Studies also show that the diphosphate group forms stabilizing interactions with several amino acid residues (two lysines, an arginine, a histidine, and a tyrosine) in the enzyme's active site, making it a weaker base and thus a better leaving group.
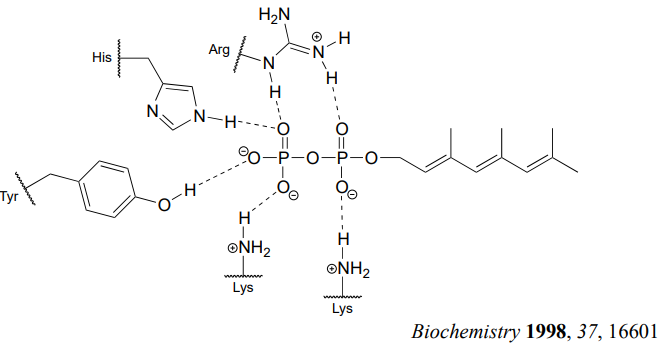
Is protein prenylation an \(S_N1\) or \(S_N2\) reaction? In other words, to what extent does the nucleophile displace, or 'push' the leaving group off, or to what extent does the leaving group leave on its own, without a 'push' from the nucleophile? Along the same lines, to what extent does a positive charge develop on the carbon center (development of a full positive charge implies an \(S_N1\) mechanism). First, consider the electrophile: it is a primary allylic carbon, so either pathway is possible (it is relatively unhindered for \(S_N2\) attack, but could also form a resonance-stabilized carbocation intermediate in an \(S_N1\) pathway). The nucleophile is a very powerful thiolate ion, suggestive of an \(S_N2\) mechanism where a strong nucleophile actively displaces the leaving group.
In fact, experiments designed to address this very question (see problem P8.19) have provided evidence that the reaction is a mechanistic hybrid: essentially \(S_N2\), but with elements of \(S_N1\). In other words, at the transition state the electrophilic carbon takes on some degree of positive charge, but a true carbocation intermediate does not form. The take-home message here is that the \(S_N1\) and \(S_N2\) mechanistic pictures we have studied in this chapter are models, and while they are useful for learning about chemical principles and accurate for describing many substitution reactions, other reactions are not necessarily 'pure' \(S_N1\) or \(S_N2\), but actually lie somewhere in between.