
6.1: Molecular Descriptors
- Page ID
- 192626
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Molecular Similarity
Molecular similarity [1-3] is one of the most heavily exploited concepts in cheminformatics and related areas (such as medicinal chemistry and drug discovery). It is applied to multiple tasks, including similarity searching [1], property prediction [4], synthesis design [5], virtual screening [2,3,6], cluster analysis [7,8], and molecular diversity analysis [9-11]. However, because molecular similarity is a concept, not a physical observable, “measuring” molecular similarity is inherently subjective and context-dependent. There is no correct or authoritative measure of molecular similarity. As a result, various similarity measures have been proposed to quantify the degree of structural similarity between molecules. In general, these measures involve two principal components [12]:
- Molecular descriptors that represent the structures of the molecules being compared.
- Similarity coefficient (metric) used to compute a quantitative score for the degree of similarity based on the weighted values of structural descriptors.
The molecular descriptors may need to be pre-processed before the similarity calculation, using a weighting scheme that assigns differing degrees of importance to various components of molecular descriptors. For this reason, some papers list the weighting scheme as a third component of similarity measures [1,13]. While some studies [14,15] have focused on the effects of the weighting schemes upon similarity calculations, much more attention has been given to molecular descriptors and similarity coefficients. Therefore, this chapter also focuses on these two components.
Molecular descriptors
There are many molecular descriptors that capture different aspects of molecules, but they are broadly classified according to their “dimensionality” [16]. One-dimensional (1-D) descriptors include bulk properties and physicochemical parameters (e.g., log P, molecular weight, polar surface area). Two-dimensional (2-D) descriptors include structural fragments or connectivity indices derived from the 2-D representation of the molecule. Three-dimensional (3-D) descriptors, such as molecular shape, are derived from 3-D molecular structures (i.e., 3-D coordinates of the atoms in the molecule). In this chapter, we focus on 2-D molecular fingerprints, which encodes the 2-D structure of molecules. While many molecular fingerprints have been developed, we discuss two types of molecular fingerprints, structural keys and hashed fingerprints, because they are more widely used than others.
Structural keys
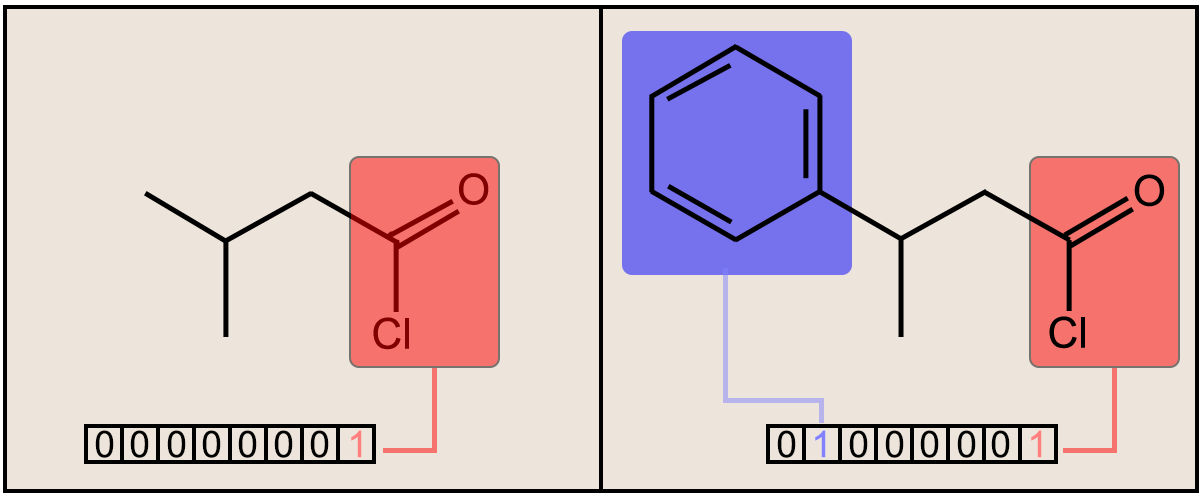
Fig. 1. (above) Two molecules are shown along with the respective bit substructures highlighted for comparison. The number of bits and designations used for this figure is simply for display and illustrative purposes. The true fingerprint would be much longer.
In structural keys, the structure of a molecule is encoded into a binary bit string (that is, a sequence of 0’s and 1’s), each bit of which corresponds to a “pre-defined” structural feature (e.g., substructure or fragment). If the molecule has a pre-defined feature, the bit position corresponding to this feature is set to 1 (ON). Otherwise, it is set to 0 (OFF). It is important to understand that structural keys cannot encode structural features that are not pre-defined in the fragment library. Examples are the MACCS keys [17,18] and PubChem Fingerprints [19].
-
MACCS keys
The MACCS (Molecular ACCess System) keys [17,18] are one of the most commonly used structural keys. They are sometimes referred to as the MDL keys, named after the company that developed them [the MDL Information Systems (now BIOVIA)]. While there are two sets of MACCS keys (one with 960 keys and the other containing a subset of 166 keys), only the shorter fragment definitions are available to the public. These 166 public keys are implemented in popular open-source cheminformatics software packages, including RDKit [20], OpenBabel [21,22], CDK [23,24], etc. The fragment definitions for the MACCS 166 keys can be found in this document:
https://github.com/rdkit/rdkit/blob/master/rdkit/Chem/MACCSkeys.py
-
PubChem fingerprints
The PubChem fingerprint [19] is a 881-bit-long structural key, which is used by PubChem for similarity searching (interactively through the PubChem Homepage or programmatically through PUG-REST). It is also used for structure neighboring, which “pre-computes” a list of similar chemical structure for each compound. This pre-computed list is accessible through the Compound Summary page (the Related Compounds and Related Compounds with Annotation sections). The fragment dictionary of the PubChem fingerprint is organized in seven sections, as described in the following document:
ftp://ftp.ncbi.nlm.nih.gov/pubchem/specifications/pubchem_fingerprints.pdf
Hashed Fingerprints
An alternative to structural keys is hashed fingerprints. Contrary to structural keys, hashed fingerprints do not require a pre-defined fragment library. Instead, they are generated by enumerating through the molecule all possible fragments that are not bigger than a certain size and then converting these fragments into numeric values using a “hash” function (https://en.Wikipedia.org/wiki/Hash_function). These numeric values can be used to indicate bit positions in the hashed fingerprints.
Hash functions are used to map data of arbitrary size to “fixed-size” values. Enumerating all possible fragments with a molecule may result in a very large number of fragments. Hashing them into values within a fixed range inevitably results in “bit collisions”, in which different fragments are converted into the same numeric value (and the same bit position). Because of this, there is no one-to-one correspondence between fragments and fingerprint bits (contrary to structural keys).
Hashed fingerprints may be further classified into topological or path-based fingerprints and circular fingerprints, according to the way by which the fragments are enumerated.
-
Path-based fingerprints
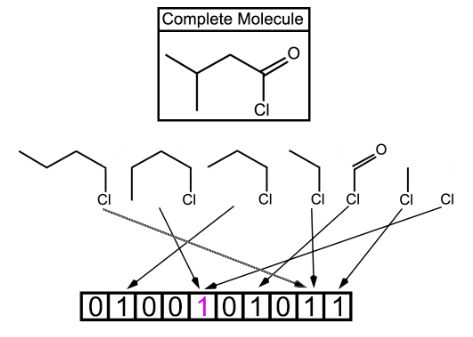
Fig. 2. Shown above is a topological fingerprint with multiple collisions between fragments. A bit collision is represented by having two or more arrows from the molecular fragments pointing to the same bit value. Starting with the chlorine atom, all of the possible fragments are shown. However in a true fingerprint, each atom could be the starting point which would allow for many more fragments than this example shows. The more bits allowed, the less likely for the bit collisions, which is represented by having two collisions due to only 10 bits being used.
In this type of fingerprints, fragments of the molecule are generated by following a (usually linear) path up to a certain number of bonds within the molecule. The most well-known example of path-based fingerprints is the Daylight fingerprint [25,26].
-
Circular fingerprints
Circular fingerprints are generated by considering the “circular” environment of each atom up to a given “radius” or “diameter”. Examples of circular fingerprints are extended-connectivity fingerprints (ECFPs) [27]. ECFPs are generated using a variant of the Morgan algorithm [28], which is a method for solving the molecular isomorphism problem (i.e., how to identify identical molecules that have different atom numberings). Different flavors of ECFPs may be generated by selecting different maximum diameter of the circular atom neighborhood and they are referred to as ECFP2, ECFP4, ECFP6, etc., where the digit at the end indicates the maximum diameter value employed to generate the fingerprint. The most commonly used ones are ECFP4 and ECFP6.
Another example of circular fingerprints is functional-class fingerprints (FCFPs) [27], which are a variation of ECFPs. FCFPs are further abstracted in that FCFPs encodes atom’s roles (not atoms). At the initial stage of FCFP generation, each atom in the molecule is assigned a special code that represents one of the atom roles (e.g., hydrogen-bond acceptor and donor, negatively or positively ionizable, aromatic, and halogen), and these codes (not the atoms) are used to generate FCFPs, through the same process as ECFPs.
References
- Willett P, Barnard JM, Downs GM: Chemical similarity searching. J Chem Inf Comput Sci 1998, 38:983-996.
- Cereto-Massague A, Ojeda MJ, Valls C, Mulero M, Garcia-Vallve S, Pujadas G: Molecular fingerprint similarity search in virtual screening. Methods 2015, 71:58-63.
- Muegge I, Mukherjee P: An overview of molecular fingerprint similarity search in virtual screening. Expert Opin Drug Discov 2016, 11:137-148.
- Brown RD, Martin YC: Use of structure Activity data to compare structure-based clustering methods and descriptors for use in compound selection. J Chem Inf Comput Sci 1996, 36:572-584.
- Wipke WT, Rogers D: ARTIFICIAL-INTELLIGENCE IN ORGANIC-SYNTHESIS - SST - STARTING MATERIAL SELECTION-STRATEGIES - AN APPLICATION OF SUPERSTRUCTURE SEARCH. J Chem Inf Comput Sci 1984, 24:71-81.
- Eckert H, Bojorath J: Molecular similarity analysis in virtual screening: foundations, limitations and novel approaches. Drug Discov Today 2007, 12:225-233.
- Cruz R, Lopez N, Quintero M, Rojas G: Cluster analysis from molecular similarity matrices using a non-linear neural network. J Math Chem 1996, 20:385-394.
- Pan DH, Iyer M, Liu JZ, Li Y, Hopfinger AJ: Constructing optimum blood brain barrier QSAR models using a combination of 4D-molecular similarity measures and cluster analysis. J Chem Inf Comput Sci 2004, 44:2083-2098.
- Golbraikh A: Molecular dataset diversity indices and their applications to comparison of chemical databases and QSAR analysis. J Chem Inf Comput Sci 2000, 40:414-425.
- Klein CT, Kaiser D, Ecker G: Topological distance based 3D descriptors for use in QSAR and diversity analysis. J Chem Inf Comput Sci 2004, 44:200-209.
- Koutsoukas A, Paricharak S, Galloway W, Spring DR, Ijzerman AP, Glen RC, Marcus D, Bender A: How Diverse Are Diversity Assessment Methods? A Comparative Analysis and Benchmarking of Molecular Descriptor Space. J Chem Inf Model 2014, 54:230-242.
- Holliday JD, Hu CY, Willett P: Grouping of coefficients for the calculation of inter-molecular similarity and dissimilarity using 2D fragment bit-strings. Comb Chem High Throughput Screen 2002, 5:155-166.
- Chen X, Reynolds CH: Performance of similarity measures in 2D fragment-based similarity searching: Comparison of structural descriptors and similarity coefficients. J Chem Inf Comput Sci 2002, 42:1407-1414.
- Bath PA, Morris CA, Willett P: EFFECT OF STANDARDIZATION ON FRAGMENT-BASED MEASURES OF STRUCTURAL SIMILARITY. J Chemometr 1993, 7:543-550.
- Turner DB, Willett P, Ferguson AM, Heritage TW: Similarity Searching in Files of Three-Dimensional Structures: Evaluation of Similarity Coefficients and Standardisation Methods for Field-Based Similarity Searching. SAR and QSAR in Environmental Research 1995, 3:101-130.
- Xue L, Bajorath J: Molecular descriptors in chemoinformatics, computational combinatorial chemistry, and virtual screening. Comb Chem High Throughput Screen 2000, 3:363-372.
- Durant JL, Leland BA, Henry DR, Nourse JG: Reoptimization of MDL keys for use in drug discovery. J Chem Inf Comput Sci 2002, 42:1273-1280.
- THE KEYS TO UNDERSTANDING MDL KEYSET TECHNOLOGY. https://www.3dsbiovia.com/products/pdf/keys-to-keyset-technology.pdf. Accessed Oct. 2019.
- PubChem Substructure Fingerprint. ftp://ftp.ncbi.nlm.nih.gov/pubchem/specifications/pubchem_fingerprints.pdf. Accessed Oct. 2019.
- RDKit. https://www.rdkit.org/. Accessed October 2019.
- O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR: Open Babel: An open chemical toolbox. J Cheminformatics 2011, 3:33.
- The Open Babel Package. https://openbabel.org. Accessed October, 2019.
- Chemistry Development Kit (CDK). https://cdk.github.io/. Accessed October 2019.
- Willighagen EL, Mayfield JW, Alvarsson J, Berg A, Carlsson L, Jeliazkova N, Kuhn S, Pluskal T, Rojas-Cherto M, Spjuth O, Torrance G, Evelo CT, Guha R, Steinbeck C: The Chemistry Development Kit (CDK) v2.0: atom typing, depiction, molecular formulas, and substructure searching. J Cheminformatics 2017, 9:33.
- Daylight Theory: Fingerprints. https://www.daylight.com/dayhtml/doc/theory/theory.finger.html. Accessed October 2019.
- Daylight Fingerprints. https://www.daylight.com/meetings/summerschool01/course/basics/fp.html. Accessed October 2019.
- Rogers D, Hahn M: Extended-Connectivity Fingerprints. J Chem Inf Model 2010, 50:742-754.
- Morgan HL: GENERATION OF A UNIQUE MACHINE DESCRIPTION FOR CHEMICAL STRUCTURES-A TECHNIQUE DEVELOPED AT CHEMICAL ABSTRACTS SERVICE. Journal of Chemical Documentation 1965, 5:107-&.
Tags recommended by the template: article:topic