
9.8: Exercise 5 - Modeling an Intramolecular Rearrangement
- Page ID
- 419805

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)This exercise is designed to calculate the transition state of the thermal conversion of vinyl alcohol to acetaldehyde using a semi-empirical calculation method. Along the way it will show you how you can model a chemical reaction (in this case an intramolecular rearrangement), and using spreadsheet functionality and graphing to create a reaction profile.
- Start a new file. Build vinyl alcohol. In the Organic tab, use
to construct the carbon skeleton. Select the
oxygen and click on one of the free valences on the vinyl skeleton. If needed rotate the CO bond into the cis-configuration (i.e., bringing the alcohol hydrogen into position over the C=C double bond). Minimize the structure using
.

- Setup the following calculation: Equilibrium Geometry from Ground State in Water, with Semi-Empirical, AM1. Total charge: Neutral, Unpaired Electrons : 0. Click Submit. Save the file as vinyl alcohol, and let the calculation run
- When it has finished, go the Summary tab of the Output and record the Energy in kJ/mol in your lab notebook.
- Select Save As from the File menu and create a copy of this molecule under the name, vinyl alcohol_ts. Select Guess Transition State from the Build menu.
- Click on the C=C bond and then the C-O bond. Next, click on the O-H bond and, while holding down the SHIFT key, click on the O-H hydrogen to be transferred and then on the carbon atom to receive this hydrogen. Another electron-pushing arrow should appear along with a faint blue dotted line between the O-H hydrogen and the carbon. It should look like this.
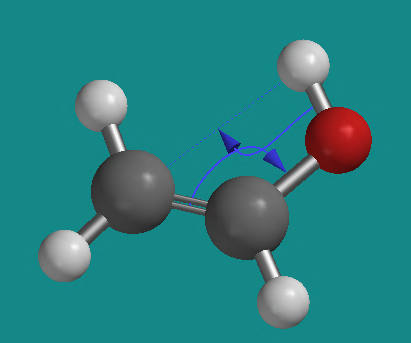
- Click on the
button at the lower right of the screen. In a few seconds your initial structure will be replaced by a calculated transition state structure. There should be a name (1_3-H-Shift_ethenol_AM1.ts (Exact)) at the lower right corner of the screen, and the structure should look like this.
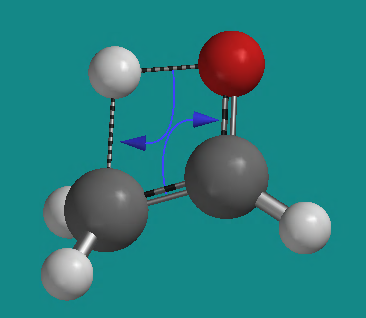
- Setup the following calculation: Calculate Transition State Geometry with Semi-empirical with AM1 in Water. Charge should be Neutral; Unpaired Electrons, 0; compute: IR. Submit the job.
- When it has finished, examine the geometry of the transition state using the Geometry functions (bond lengths and angles). Go to Display, Output and record the energy of the transition state structure.
- Select Spectra under the Display menu. Use the
and
to display the calculated IR and frequency table. Animate the vibrational mode corresponding to the “imaginary” (i) frequency. You might click the
button and change the Steps to 30 to slow down the animation.
The idea is this: By now I am sure you are familiar with a potential energy well showing how the energy of a bond varies as a function of internuclear distance. The minimum of this well represents the equilibrium bond length. Additionally, you know that the tie-lines across this potential well represent vibrational modes of the bond. Now a similar tie line across the potential energy curve of a reaction profile, near where the maximum in the curve represents the transition state of the molecule. We can imagine, and calculate a value for, such a vibrational mode associated with this idea. This calculated "imaginary" vibration is typically denoted with an i, and models the dynamics of the molecule as it traverses over the top of the activation energy barrier through the transition state structure.
Notice in the animation that the C=C double bond lengthens as the hydrogen moves toward the carbon, and that the carbon accepting this hydrogen shifts from sp2 toward sp3 geometry. Notice also that the C-O bond shortens with the hydride shift as it starts to become a C=O bond. And also that the forming carbonyl carbon shows the shift from sp3 to sp2 geometry. That is, this one calculated imaginary vibration shows the structural changes in the transition state and across the top of the activation energy barrier in a reaction profile. Stop the animation and close the Spectrum window.
- Build acetaldehyde as a New file. Set up the same Equilibrium Geometry calculation as you did for vinyl alcohol (Semi-empirical, AM1, Water). Save the file and Submit the calculation. Record the energy as you did for vinyl alcohol and the transition state. Close your molecule.

Creating the Reaction Profile:
- Open your vinyl alcohol molecule made and optimized in steps 4.8.1-4.8.2. From the File menu, select Save-as and save the file as Reaction Profile. Select Spreadsheet from the Display menu. A spreadsheet appears on the screen with one entry. This entry is your vinyl alcohol molecule. Double-click on the M001 entry and change the name to Vinyl Alcohol.
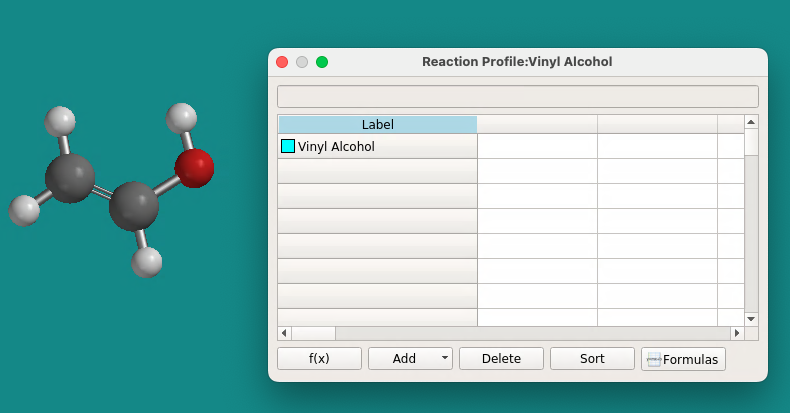
- Now open the transition state molecule created and optimized in steps 4.8.4-4.8.7 above. Notice that this molecule opens in its own window (you can see the tabs at the lower left of the Spartan screen). Use Select All from the File menu to highlight the molecule (or right click on the molecule and select all). Use Copy from the File menu (or right mouse click – Copy) to copy the molecule to the Spartan clipboard. Return to the vinyl alcohol/spreadsheet screen by clicking on its tab. Click once on the cell immediately under the vinyl alcohol entry. Right-click on that same cell and paste the transition state into the spreadsheet. Change the name to Transition State.
- Open and paste your acetaldehyde molecule into the third spreadsheet cell. At this point your spreadsheet will look like this.

- Open the Setup Calculations box for each of the three molecules in the spreadsheet and confirm that the desired calculation is correct for each. If it is not, correct them. It should be Equilibrium Geometry in water, with semi-empirical, AM1 for vinyl alcohol and acetaldehyde; and, Transition State Geometry in water with AM1 for transition state molecule. Click OK (not Submit) to close the Setup Calculations box each time.
- Click the Add button in the spreadsheet window. Select Energy (kJ/mol) from the list of choices. Click OK. You will notice a new Energy column appear in the spreadsheet, with Pending next to each molecule.
- Return to the Setup Calculations box. Click Submit. The calculations will run and when complete the spreadsheet will show the aqueous energies for all three entries.

- Select Plots from the Display menu. Click on the
button and highlight Energy in the window that appears. Click Create. A plot appears in a window to the right of the molecule screen. Click
and select Smooth from the Curve options. Add a Title, and click Done. Click
, and the plot, showing the reaction profile (Energy vs molecule) will move to the main screen. You can use the Animation bar,
, to step through the profile, showing the structure at each point in the process. Record images of the reaction profile in your lab notebook. Use the energy values from the spreadsheet to calculate the activation energy and \( \Delta H\) for the reaction.

This section of your lab notebook should include:
- The equilibrium geometries energies of the initial, transition, and final states
- Images of all reaction profile plots
- Calculation of the activation energy and \( \Delta H\) of the tautomerization reaction
(See your ELN template for details)