
13.8: Rotational Spectra of Polyatomic Molecules
- Page ID
- 63747
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)As discussed previously, the Schrödinger equation for the angular motion of a rigid (i.e., having fixed bond length \(R\)) diatomic molecule is
\[\dfrac{\hbar^2}{2 μ} \left[ \dfrac{1}{R^2 \sin θ} \dfrac{∂}{∂θ} \left(\sin θ \dfrac{∂}{∂θ} \right) + \dfrac{1}{R^2 \sin^2 θ} \dfrac{∂^2}{∂φ^2} \right] |ψ \rangle = E | ψ \rangle \nonumber \]
or
\[ \dfrac{L^2}{2 μ R^2 } | ψ \rangle = E | ψ\rangle \nonumber \]
The Hamiltonian in this problem contains only the kinetic energy of rotation; no potential energy is present because the molecule is undergoing unhindered "free rotation". The angles \(θ\) and \(φ\) describe the orientation of the diatomic molecule's axis relative to a laboratory-fixed coordinate system, and \(μ\) is the reduced mass of the diatomic molecule
\[ μ=\dfrac{m_1m_2}{m_1+m_2} \nonumber \]
The eigenvalues corresponding to each eigenfunction are straightforward to find because \(H_{rot}\) is proportional to the \(L^2\) operator whose eigenvalues have already been determined. The resultant rotational energies are given as:
\[E_J= \dfrac{\hbar^2J(J+1)}{2μR^2} = B J(J+1) \label{Ediatomic} \]
and are independent of \(M\). \(B\) is the rotational constant. Thus each energy level is labeled by \(J\) and is \(2J+1\)-fold degenerate (because \(M\) ranges from \(-J\) to \(J\)). The rotational energy in Equation \(\ref{Ediatomic}\) can be expressed in terms of the moment of inertia \(I\)
\[I =\sum_i m_i R_i^2 \label{Idiatomic} \]
where \(m_i\) is the mass of the \(i^{th}\) atom and \(R\) is its distance from the center of mass of the molecule. This moment of inertia replaces \(μR^2\) in the denominator of Equation \(\ref{Ediatomic}\):
\[E_J= \dfrac{\hbar^2J(J+1)}{2I} = B J(J+1) \label{Ediatomic2} \]
Rotation of Polyatomic Molecules
In contrast to diatomic molecules (Equation \ref{Idiatomic}), the rotational motions of polyatomic molecules in three dimensions are characterized by multiple moments of inertia. typically reflected in an \(3 \times 3\) inertia tensor. It is common in rigid body mechanics to express in these moments of inertia in lab-based Cartesian coordinates via a notation that explicitly identifies the \(x\), \(y\), and \(z\) axes such as \(I_{xx}\) and \(I_{xy}\), for the components of the inertia tensor.
\[I =\begin{bmatrix}I_{xx}&I_{xy}&I_{xz}\\I_{yx}&I_{yy}&I_{yz}\\I_{zx}&I_{zy}&I_{zz}\end{bmatrix} \label{inertiamatrix} \]
The components of this tensor can be assembled into a matrix given by
\[ I_{xx}=\sum _{k=1}^{N}m_{k}(y_{k}^{2}+z_{k}^{2}) \nonumber \]
\[ I_{yy}=\sum _{k=1}^{N}m_{k}(x_{k}^{2}+z_{k}^{2}) \nonumber \]
\[ I_{zz}=\sum _{k=1}^{N}m_{k}(x_{k}^{2}+y_{k}^{2}) \nonumber \]
\[ I_{yx}=I_{xy}=-\sum _{k=1}^{N}m_{k}x_{k}y_{k} \nonumber \]
\[ I_{zx}=I_{xz}=-\sum _{k=1}^{N}m_{k}x_{k}z_{k} \nonumber \]
\[ I_{zy}=I_{yz}=-\sum _{k=1}^{N}m_{k}y_{k}z_{k}. \nonumber \]
The rotational motions of polyatomic molecules are characterized by moments of inertia that are defined in a molecule based coordinates with axes that are labeled \(a\), \(b\), and \(c\). Measured in the body frame the inertia matrix (Equation \(\ref{inertiamatrix}\)) is a constant real symmetric matrix, which can be decomposed into a diagonal matrix, given by
\[ I =\left(\begin{array}{ccc}I_{a}&0&0\\0&I_{b}&0\\0&0&I_{c}\end{array}\right) \nonumber \]
These labels are assigned so that \(I_c\) is the largest principal moment of inertia with an order of the three moments set as
\[ I_a<I_b<I_c \nonumber \]
The rotational kinetic energy operator for a rigid non-linear polyatomic molecule is then expressed as
\[H_{rot} = \dfrac{J_a^2}{2I_a} + \dfrac{J_b^2}{2I_b} + \dfrac{J_c^2}{2I_c} \label{genKE} \]
The components of the quantum mechanical angular momentum operators along the three principal axes are:
\[ \begin{align} J_a &= -i\hbar \cos χ \left[\cot θ \dfrac{∂}{∂χ} - (\sin θ )^{-1} \dfrac{∂}{∂φ} \right] - -i\hbar \sin χ \dfrac{∂}{∂θ} \\[4pt] J_b &= i\hbar \sin χ \left[\cot θ \dfrac{∂}{∂χ} - (\sin θ )^{-1} \dfrac{∂}{∂φ} \right] - -i\hbar \cos χ \dfrac{∂}{∂θ} \\[4pt] J_c &= - \dfrac{ih ∂}{∂χ} \end{align} \nonumber \]
The angles \(θ\), \(φ\), and \(χ\) are the Euler angles needed to specify the orientation of the rigid molecule relative to a laboratory-fixed coordinate system. The corresponding square of the total angular momentum operator \(J^2\) can be obtained as
\[ \begin{align} J^2 &= J_a^2 + J_b^ 2 + J_c^2 \\[4pt] & = - \dfrac{∂^2}{∂θ^2} - \cot θ \dfrac{∂}{∂θ} - \left(\dfrac{1}{\sin θ} \right) \left( \dfrac{∂^2}{∂φ^2} + \dfrac{∂^2}{∂χ^2} - 2 \cos θ \dfrac{∂^2}{∂φ∂χ} \right) \end{align} \nonumber \]
and the component along the lab-fixed \(Z\) axis is
\[J_Z = - ih \dfrac{∂}{∂φ}. \nonumber \]
Spherical Tops
When the three principal moment of inertia values are identical, the molecule is termed a spherical top. In this case, the total rotational energy Equation \(\ref{genKE}\) can be expressed in terms of the total angular momentum operator \(J^2\)
\[H_{rot} = \dfrac{J^2}{2I} \nonumber \]
As a result, the eigenfunctions of \(H_{rot}\) are those of \(J^2\) (and \(J_a\) as well as \(J_Z\) both of which commute with \(J_2\) and with one another; \(J_Z\) is the component of \(J\) along the lab-fixed Z-axis and commutes with \(J_a\) because
\[J_Z = - ih \dfrac{∂}{∂φ} \nonumber \]
and
\[J_a = - ih \dfrac{∂}{∂χ} \nonumber \]
act on different angles. The energies associated with such eigenfunctions are
\[E(J,K,M) = \dfrac{\hbar^2 J(J+1)}{2I^2} \nonumber \]
for all K (i.e., J a quantum numbers) ranging from -J to J in unit steps and for all M (i.e., J Z quantum numbers) ranging from -J to J. Each energy level is therefore \((2J + 1)^2\) degenarate because there are \(2J + 1\) possible K values and \(2J + 1\) possible M values for each J. The eigenfunctions of \(J^2\), \(J_Z\) and \(J_a\), \(|J,M,K>\) are given in terms of the set of rotation matrices \(D_{J,M,K}\) :
\[|J,M,K \rangle = \sqrt{ \dfrac{2J + 1}{8 π^2}} D^* _{J,M,K} ( θ , φ , χ ) \nonumber \]
which obey
\[J^2 |J,M,K \rangle = \hbar^2 J(J+1) | J,M,K \rangle \nonumber \]
\[J_a |J,M,K \rangle = \hbar K | J,M,K \rangle \nonumber \]
\[J_Z |J,M,K \rangle = \hbar M | J,M,K \rangle \nonumber \]
Symmetric Tops
Symmetrical tops are molecules with two rotational axes that have the same inertia and one unique rotational axis with a different inertia. Symmetrical tops can be divided into two categories based on the relationship between the inertia of the unique axis and the inertia of the two axes with equivalent inertia. If the unique rotational axis has a greater inertia than the degenerate axes the molecule is called an oblate symmetrical top (Figure 13.8.1 ). If the unique rotational axis has a lower inertia than the degenerate axes the molecule is called a prolate symmetrical top. For simplification think of these two categories as either frisbees for oblate tops or footballs for prolate tops.
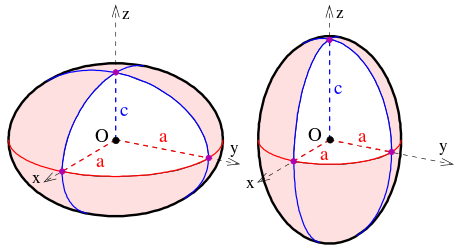
Again, the rotational kinetic energy, which is the full rotational Hamiltonian, can be written in terms of the total rotational angular momentum operator \(J^2\) and the component of angular momentum along the axis with the unique principal moment of inertia.
For prolate tops, Equation \(\ref{genKE}\) becomes
\[H_{rot} = \dfrac{J^2}{2I} + J_a^2 \left( \dfrac{1}{2I_a} - \dfrac{1}{2I} \right) \nonumber \]
For oblate tops, Equation \(\ref{genKE}\) becomes
\[H_{rot} = \dfrac{J^2}{2I} + J_c^2 \left( \dfrac{1}{2I_c} - \dfrac{1}{2I} \right) \nonumber \]
As a result, the eigenfunctions of \(H_{rot}\) are those of \(J^2\) and \(J_a\) or \(J_c\) (and of \(J_Z\)), and the corresponding energy levels.
The energies for prolate tops are
\[E(J,K,M) = \dfrac{h^2 J(J+1)}{2I^2} + h^2 K^2 \left( \dfrac{1}{2I_a} - \dfrac{1}{2I} \right) \nonumber \]
and the energies for oblate tops are
\[E(J,K,M) = \dfrac{h^2 J(J+1)}{2I 2} + h^2 K^2 \left( \dfrac{1}{2I_c} - \dfrac{1}{2I} \right) \nonumber \]
again for K and M (i.e., \(J_a\) or \(J_c\) and \(J_Z\) quantum numbers, respectively) ranging from \(-J\) to \(J\) in unit steps. Since the energy now depends on K, these levels are only \(2J + 1\) degenerate due to the \(2J + 1\) different \(M\) values that arise for each \(J\) value. The eigenfunctions \(|J, M,K>\) are the same rotation matrix functions as arise for the spherical-top case.
Asymmetric Tops
The rotational eigenfunctions and energy levels of a molecule for which all three principal moments of inertia are distinct (a asymmetric top) can not easily be expressed in terms of the angular momentum eigenstates and the \(J\), \(M\), and \(K\) quantum numbers. However, given the three principal moments of inertia \(I_a\), \(I_b\), and \(I_c\), a matrix representation of each of the three contributions to the general rotational Hamiltonian in Equation \(\ref{genKE}\) can be formed within a basis set of the \(\{|J, M, K \rangle\}\) rotation matrix functions. This matrix will not be diagonal because the \(|J, M, K \rangle\) functions are not eigenfunctions of the asymmetric top \(H_{rot}\). However, the matrix can be formed in this basis and subsequently brought to diagonal form by finding its eigenvectors {C n, J,M,K } and its eigenvalues \(\{E_n\}\). The vector coefficients express the asymmetric top eigenstates as
\[\psi_n ( θ , φ , χ ) = \sum_{J, M, K} C_{n, J,M,K} |J, M, K \rangle \nonumber \]
Because the total angular momentum \(J^2\) still commutes with \(H_{rot}\), each such eigenstate will contain only one J-value, and hence \(Ψ_n\) can also be labeled by a \(J\) quantum number:
\[\psi _{n,J} ( θ , φ , χ ) = \sum_{M, K} C_{n, J,M,K} |J, M, K \rangle \nonumber \]
To form the only non-zero matrix elements of \(H_{rot}\) within the \(|J, M, K\rangle\) basis, one can use the following properties of the rotation-matrix functions:
\[\langle j, \rangle = \langle j, \rangle = 1/2 <j, \rangle = h 2 [ J(J+1) - K 2 ], \nonumber \]
\[ \langle j, \rangle = h^2 K^2 \nonumber \]
\[ \langle j \rangle = - \langle j \rangle = h^2 [J(J+1) - K(K ± 1)] 1/2 [J(J+1) -(K ± 1)(K ± 2)] 1/2 \langle j \rangle = 0 \nonumber \]
Each of the elements of \(J_c^2\), \(J_a^2\), and \(J_b^2\) must, of course, be multiplied, respectively, by \(1/2I_c\), \(1/2I_a\), and \(1/2I_b\) and summed together to form the matrix representation of \(H_{rot}\). The diagonalization of this matrix then provides the asymmetric top energies and wavefunctions.