
High Performance Liquid Chromatography
- Page ID
- 202040
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)High Performance Liquid Chromotagraphy (HPLC) is an analytical technique used for the separation of compounds soluble in a particular solvent.
History of HPLC
Liquid chromatography was initially discovered as an analytical technique in the early twentieth century and was first used as a method of separating colored compounds. This is where the name chromatography chroma means color, graphy means writing, was derived. A Russian botanist named Mikhail S. Tswett used a rudimentary form of chromatographic separation to purify mixtures of plant pigments into the pure constituents. He separated the pigments based on their interaction with a stationary phase, which is essential to any chromatographic separation. The stationary phase he used was powdered chalk and aluminia, the mobile phase in his separation was the solvent. After the solid stationary phase was packed into a glass column (essentially a long, hollow, glass tube) he poured the mixture of plant pigments and solvent in the top of the column. He then poured additional solvent into the column until the samples were eluted at the bottom of the column. The result of this process most crucial to his investigation was that the plant pigments separated into bands of pure components as they passed through the stationary phase. Modern high performance liquid chromatography or HPLC has its roots in this separation, the first form of liquid chromatography. The chromatographic process has been significantly improved over the last hundred years, yielding greater separation efficiency, versatility and speed.
Affinities for Mobile and Stationary Phases
All chromatographic separations, including HPLC operate under the same basic principle; every compound interacts with other chemical species in a characteristic manner. Chromatography separates a sample into its constituent parts because of the difference in the relative affinities of different molecules for the mobile phase and the stationary phase used in the separation.
Distribution Constant
All chemical reactions have a characteristic equilibrium constant. For the reaction
\[ A_{aq} + B_s \rightleftharpoons AB_s \label{1}\]
There is a chemical equilibrium constant Keq that dictates what percentage of compound A will be in solution and what percentage will be bound to the stationary compound B. During a chromatographic separation, there is similar relationship between compound A and the solvent, or mobile phase, C. This will yield an overall equilibrium equation which dictates the quantity of A that will be associated with the stationary phase and the quantity of A that will be associated with the mobile phase.
\[ A_{mobile} \rightleftharpoons A_{stationary} \label{2}\]
The equilibrium between the mobile phase and stationary phase is given by the constant Kc.
\[ K_c = \dfrac{(a_A )_S}{(a_A )_M} \approx \dfrac{c_S}{c_M} \label{3}\]
Where Kc, the distribution constant, is the ratio of the activity of compound A in the stationary phase and activity of compound A in the mobile phase. In most separations, which contain low concentrations of the species to be separated, the activity of A in each is approximately equal to the concentration of A in that state. The distribution constant indicates the amount of time that compound A spends adsorbed to the stationary phase as the opposed to the amount of time A spends solvated by the mobile phase. This relationship determines the amount of time it will take for compound A to travel the length of the column. The more time A spends adsorbed to the stationary phase, the more time compound A will take to travel the length of the column. The amount of time between the injection of a sample and its elution from the column is known as the retention time; it is given the symbol tR.
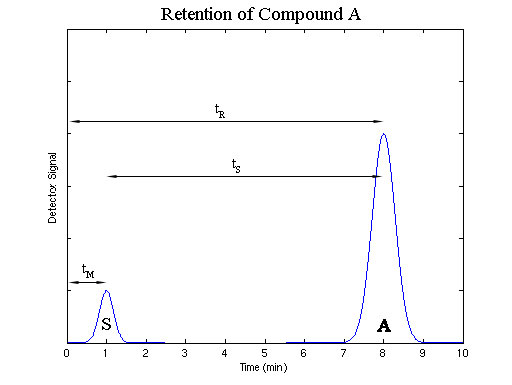
The amount of time required for a sample that does not interact with the stationary phase, or has a Kc equal to zero, to travel the length of the column is known as the void time, tM. No compound can be eluted in less than the void time.
Retention Factor
Since Kc is a factor that is wholly dependent on a particular column and solvent flow rate, a quantitative measure of the affinity of a compound for a particular set of mobile and stationary phases that does not depend on the column geometry is useful. The retention factor, k, can be derived from Kc and is independent of the column size and the solvent flow rate.
\[ k_C = \dfrac{K_C V_S }{V_M } \label{4}\]
The retention factor is calculated by multiplying the distribution constant by the volume of stationary phase in the column and dividing by the volume of mobile phase in the column.
Selectivity
In order to separate two compounds, their respective retention factors must be different, otherwise both compounds would be eluted simultaneously; the selectivity factor is the ratio of the retention factors.
\[ \alpha = \dfrac{k_B }{k_A} \label{5}\]
Where B is the compound that is retained more strongly by the column and A is the compound with the faster elution time.
Band Broadening
As a compound passes through the column it slowly diffuses away from the initial injection band, which is the area of greatest concentration. The initial, narrow, band that contained all of the sample becomes broader the longer the analyte remains in the column. This band broadening increases the time required for complete elution of a particular compound and is generally undesirable. It must be minimized so that overly broad elution bands do not overlap with one another. We will see how this is measured quantitatively when we discuss peak resolution momentarily.
Separation Efficiency
The overriding purpose of a chromatographic separation is just that, to separate two or more compounds contained in solution. In analytical chemistry, a quantitative metric of every experimental parameter is desired, and so separation efficiency is measured in plates. The concept of plates as a separation metric arose from the original method of fractional distillation, where compounds were separated based on their volatilities through many simultaneous simple distillations, each simple distillation occurred on one of many distillation plates. In chromatography, no actual plates are used, but the concept of a theoretical plate, as a distinct region where a single equilibrium is maintained, remains. In a particular liquid chromatographic separation, the number of theoretical plates and the height equivalent to a theoretical plate (HETP) are related simply by the length of the column
\[ N = \dfrac{L}{H} \label{6}\]
Where N is the number of theoretical plates, L is the length of the column, and H is the height equivalent to a theoretical plate. The plate height is given by the variance (standard deviation squared) of an elution peak divided by the length of the column.
\[ H = \dfrac{\sigma ^2}{L} \label{7}\]
The standard deviation of an elution peak can be approximated by assuming that a Gaussian elution peak is roughly triangular, in that case the plate height can be given by the width of the elution peak squared times the length of the column over the retention time of the that peak squared times 16.
\[ H = \dfrac{LW^2 }{16t_R^2} \label{8}\]
Using the relationship between plate height and number of plates, the number of plates can also be found in terms of retention time and peak width.
\[ N = 16 \left( \dfrac{t_R}{W} \right)^2\label{9} \]
In order to optimize separation efficiency, it is necessary in maximize the number of theoretical plates, which requires reducing the plate height. The plate height is related to the flow rate of the mobile phase, so for a fixed set of mobile phase, stationary phase, and analytes; separation efficiency can be maximized by optimizing flow rate as dictated by the van Deemter equation.
\[ H = A + \dfrac{B}{v} + Cv \label{10}\]
The three constants in the van Deemter equation are factors that describe possible causes of band broadening in a particular separation. \(A\) is a constant which represents the different possible paths that can be taken by the analyte through the stationary phase, it decreases if the packing of the column is kept as small as possible. \(B\) is a constant that describes the longitudinal diffusion that occurs in the system. \(C\) is a constant that describes the rate of adsorption and desorption of the analyte to the stationary phase. \(A\), \(B\) and \(C\) are constant for any given system (with constant analyte, stationary phase, and mobile phase), so flow rate must be optimized accordingly. If the flow rate is too low, the longitudinal diffusion factor (\(\dfrac{B}{v}\)) will increase significantly, which will increase plate height. At low flow rates, the analyte spends more time at rest in the column and therefore longitudinal diffusion in a more significant problem. If the flow rate is too high, the mass transfer term (\(Cv\)) will increase and reduce column efficiency. At high flow rates the adsorption of the analyte to the stationary phase results in some of the sample lagging behind, which also leads to band broadening.
Resolution
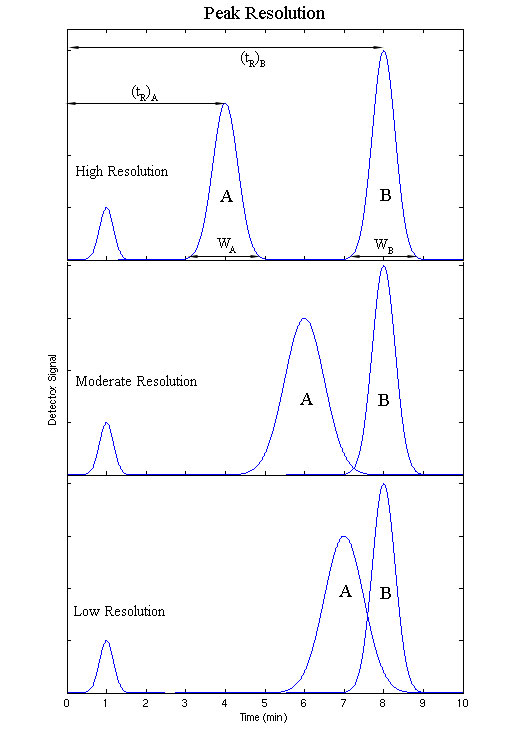
The resolution of a elution is a quantitative measure of how well two elution peaks can be differentiated in a chromatographic separation. It is defined as the difference in retention times between the two peaks, divided by the combined widths of the elution peaks.
\[ R_S = \dfrac{2\left[ {\left( {t_R } \right)_B - \left( {t_R } \right)_A } \right]}{W_B + W_A} \label{11}\]
Where B is the species with the longer retention time, and tR and W are the retention time and elution peak width respectively. If the resolution is greater than one, the peaks can usually be differentiated successfully.
HPLC as a solution to efficiency problems
While all of these basic principles hold true for all chromatographic separations, HPLC was developed as method to solve some of the shortcomings of standard liquid chromatography. Classic liquid chromatography has several severe limitations as a separation method. When the solvent is driven by gravity, the separation is very slow, and if the solvent is driven by vacuum, in a standard packed column, the plate height increases and the effect of the vacuum is negated. The limiting factor in liquid chromatography was originally the size of the column packing, once columns could be packed with particles as small as 3 µm, faster separations could be performed in smaller, narrower, columns. High pressure was required to force the mobile phase and sample through these new columns, and previously unneeded apparatus was required to maintain reproducibility of results in this new instruments. The use of high pressures in a narrow column allowed for a more effective separation to be achieved in much less time than was required for previous forms of liquid chromatography.
Apparatus
Specialized apparatus is required for an HPLC separation because of the high pressures and low tolerances under which the separation occurs. If the results are to be reproducible, then the conditions of the separation must also be reproducible. Thus HPLC equipment must be of high quality; it is therefore expensive.
Solvent
The mobile phase, or solvent, in HPLC is usually a mixture of polar and non-polar liquid components whose respective concentrations are varied depending on the composition of the sample. As the solvent is passed through a very narrow bore column, any contaminants could at worst plug the column, or at the very least add variability to the retention times during repeated different trials. Therefore HPLC solvent must be kept free of dissolved gases, which could come out of solution mid-separation, and particulates.
Column
In the HPLC column, the components of the sample separate based on their differing interactions with the column packing. If a species interacts more strongly with the stationary phase in the column, it will spend more time adsorbed to the column's adsorbent and will therefore have a greater retention time. Columns can be packed with solids such as silica or alumina; these columns are called homogeneous columns. If stationary phase in the column is a liquid, the column is deemed a bonded column. Bonded columns contain a liquid stationary phase bonded to a sold support, which is again usually silica or alumina. The value of the constant C described in the van Deemter equation is proportional, in HPLC, to the diameter of the particles that constitute the column's packing material.
Pump
The HPLC pump drives the solvent and sample through the column. To reduce variation in the elution, the pump must maintain a constant, pulse free, flow rate; this is achieved with multi-piston pumps. The presence of two pistons allows the flow rate to be controlled by one piston as the other recharges. A syringe pump can be used for even greater control of flow rate; however, the syringe pump is unable to produce as much pressure as a piston pump, so it cannot be used in all HPLC applications.
Detector
The HPLC detector, located at the end of the column, must register the presence of various components of the sample, but must not detect the solvent. For that reason there is no universal detector that works for all separations. A common HPLC detector is a UV absorption detector, as most medium to large molecules absorb UV radiation. Detectors that measure fluorescence and refractive index are also used for special applications. A relatively new development is the combination of an HPLC separation with an NMR detector. This allows the pure components of the sample to be identified and quantified by nuclear magnetic resonance after having been separated by HPLC, in one integrated process.
Technique
Normal Phase vs. Reverse Phase
If the stationary phase is more polar than the mobile phase, the separation is deemed normal phase. If the stationary phase is less polar than the mobile phase, the separation is reverse phase. In reverse phase HPLC the retention time of a compound increases with decreasing polarity of the particular species. The key to an effective and efficient separation is to determine the appropriate ratio between polar and non-polar components in the mobile phase. The goal is for all the compounds to elute in as short a time as possible, while still allowing for the resolution of individual peaks. Typical columns for normal phase separation are packed with alumina or silica. Alkyl, aliphatic or phenyl bonded phases are typically used for reverse phase separation.
Gradient Elution vs. Isocratic Elution
If the composition of the mobile phase remains constant throughout the HPLC separation, the separation is deemed an isocratic elution. Often the only way to elute all of the compounds in the sample in a reasonable amount of time, while still maintaining peak resolution, is to change the ratio of polar to non-polar compounds in the mobile phase during the sample run. Known as gradient chromatography, this is the technique of choice when a sample contains components of a wide range of polarities. For a reverse phase gradient, the solvent starts out relatively polar and slowly becomes more non-polar. The gradient elution offers the most complete separation of the peaks, without taking an inordinate amount of time. A sample containing compounds of a wide range of polarities can be separated by a gradient elution in a shorter time period without a loss of resolution in the earlier peaks or excessive broadening of later peaks. However, gradient elution requires more complex and expensive equipment and it is more difficult to maintain a constant flow rate while there are constant changes in mobile phase composition. Gradient elution, especially at high speeds, brings out the limitations of lower quality experimental apparatus, making the results obtained less reproducible in equipment already prone to variation. If the flow rate or mobile phase composition fluctuates, the results will not be reproducible.
Applications
HPLC can be used in both qualitative and quantitative applications, that is for both compound identification and quantification. Normal phase HPLC is only rarely used now, almost all HPLC separation can be performed in reverse phase. Reverse phase HPLC (RPLC) is ineffective in for only a few separation types; it cannot separate inorganic ions (they can be separated by ion exchange chromatography). It cannot separate polysaccharides (they are too hydrophilic for any solid phase adsorption to occur), nor polynucleotides (they adsorb irreversibly to the reverse phase packing). Lastly, incredibly hydrophobic compounds cannot be separated effectively by RPLC (there is little selectivity). Aside from these few exceptions, RPLC is used for the separation of almost all other compound varieties. RPLC can be used to effectively separate similar simple and aromatic hydrocarbons, even those that differ only by a single methylene group. RPLC effectively separates simple amines, sugars, lipids, and even pharmaceutically active compounds. RPLC is also used in the separation of amino acids, peptides, and proteins. Finally RPLC is used to separate molecules of biological origin. The determination of caffeine content in coffee products is routinely done by RPLC in commercial applications in order to guarantee purity and quality of ground coffee. HPLC is a useful addition to an analytical arsenal, especially for the separation of a sample before further analysis.
References
- Skoog, D.; Holler, F.; Crouch, S. Principles of Instrumental Analysis 2007
- Swadesh, J.K. HPLC: Practical and Industrial Applications 2001
- Waters Corporation: History of Chromatography (accessed March 3, 2008)
Contributors
- Matthew Barkovich (UCD)