
7.3: Implementing the Sampling Plan
- Page ID
- 220715

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Implementing a sampling plan usually involves three steps: physically removing the sample from its target population, preserving the sample, and preparing the sample for analysis. Except for in situ sampling, we analyze a sample after we have removed it from its target population. Because sampling exposes the target population to potential contamination, our sampling device must be inert and clean.
Once we remove a sample from its target population, there is a danger that it will undergo a chemical or physical change before we can complete its analysis. This is a serious problem because the sample’s properties will no longer e representative of the target population. To prevent this problem, we often preserve samples before we transport them to the laboratory for analysis. Even when we analyze a sample in the field, preservation may still be necessary.
The initial sample is called the primary or gross sample, and it may be a single increment drawn from the target population or a composite of several increments. In many cases we cannot analyze the gross sample without first preparing the sample for analyze by reducing the sample’s particle size, by converting the sample into a more readily analyzable form, or by improving its homogeneity.
Although you may never work with the specific samples highlighted in this section, the case studies presented here may help you in envisioning potential problems associated with your samples.
Solutions
There are many good examples of solution samples: commercial solvents; beverages, such as milk or fruit juice; natural waters, including lakes, streams, seawater, and rain; bodily fluids, such as blood and urine; and, suspensions, such as those found in many oral medications. Let’s use the sampling of natural waters and wastewaters as a case study in how to sample a solution.
Sample Collection
The chemical composition of a surface water—such as a stream, river, lake, estuary, or ocean—is influenced by flow rate and depth. Rapidly flowing shallow streams and rivers, and shallow (<5 m) lakes usually are well mixed and show little stratification with depth. To collect a grab sample we submerge a capped bottle below the surface, remove the cap and allow the bottle to fill completely, and replace the cap. Collecting a sample this way avoids the air–water interface, which may be enriched with heavy metals or contaminated with oil [Duce, R. A.; Quinn, J. G. Olney, C. E.; Piotrowicz, S. R.; Ray, S. J.; Wade, T. L. Science 1972, 176, 161–163].
Slowly moving streams and rivers, lakes deeper than five meters, estuaries, and oceans may show substantial stratification with depth. Grab samples from near the surface are collected as described above, and samples at greater depths are collected using a sample bottle lowered to the desired depth (Figure \(\PageIndex{1}\)).

Wells for sampling groundwater are purged before we collect samples because the chemical composition of water in a well-casing may differ significantly from that of the groundwater. These differences may result from contaminants introduced while drilling the well or by a change in the groundwater’s redox potential following its exposure to atmospheric oxygen. In general, a well is purged by pumping out a volume of water equivalent to several well-casing volumes or by pumping until the water’s temperature, pH, or specific conductance is constant. A municipal water supply, such as a residence or a business, is purged before sampling because the chemical composition of water standing in a pipe may differ significantly from the treated water supply. Samples are collected at faucets after flushing the pipes for 2-3 minutes.
Samples from municipal wastewater treatment plants and industrial discharges often are collected as a 24-hour composite. An automatic sampler periodically removes an individual grab sample, adding it to those collected previously. The volume of each sample and the frequency of sampling may be constant, or may vary in response to changes in flow rate.
Sample containers for collecting natural waters and wastewaters are made from glass or plastic. Kimax and Pyrex brand borosilicate glass have the advantage of being easy to sterilize, easy to clean, and inert to all solutions except those that are strongly alkaline. The disadvantages of glass containers are cost, weight, and the ease of breakage. Plastic containers are made from a variety of polymers, including polyethylene, polypropylene, polycarbonate, polyvinyl chloride, and Teflon. Plastic containers are light-weight, durable, and, except for those manufactured from Teflon, inexpensive. In most cases glass or plastic bottles are used interchangeably, although polyethylene bottles generally are preferred because of their lower cost. Glass containers are always used when collecting samples for the analysis of pesticides, oil and grease, and organics because these species often interact with plastic surfaces. Because glass surfaces easily adsorb metal ions, plastic bottles are preferred when collecting samples for the analysis of trace metals.
In most cases the sample bottle has a wide mouth, which makes it easy to fill and to remove the sample. A narrow-mouth sample bottle is used if exposing the sample to the container’s cap or to the outside environment is a problem. Unless exposure to plastic is a problem, caps for sample bottles are manufactured from polyethylene. When polyethylene must be avoided, the container’s cap includes an inert interior liner of neoprene or Teflon.
Sample Preservation and Preparation
Here our concern is only with the need to prepare the gross sample by converting it into a form suitable for analysis. Some analytical methods require additional sample preparation steps, such as concentrating or diluting the analyte, or adjusting the analyte’s chemical form. We will consider these forms of sample preparation in later chapters that focus on specific analytical methods.
After removing a sample from its target population, its chemical composition may change as a result of chemical, biological, or physical processes. To prevent a change in composition, samples are preserved by controlling the sample’s pH and temperature, by limiting its exposure to light or to the atmosphere, or by adding a chemical preservative. After preserving a sample, it is safely stored for later analysis. The maximum holding time between preservation and analysis depends on the analyte’s stability and the effectiveness of sample preservation. Table \(\PageIndex{1}\) summarizes preservation methods and maximum holding times for several analytes of importance in the analysis of natural waters and wastewaters.
| analyte | preservation method | maximum holding time |
|---|---|---|
| ammonia |
cool to 4oC; add H2SO4 to pH < 2 |
28 days |
| chloride | none required | 28 days |
| metals: Cr(VI) | cool to 4oC | 24 hours |
| metals: Hg |
HNO3 to pH < 2 |
28 days |
| metals: all others |
HNO3 to pH < 2 |
6 months |
| nitrate |
none required |
48 hours |
| organochlorine pesticides |
1 mL of 10 mg/mL HgCl2 or immediate extraction with a suitable non-aqueous solvent |
7 days without extraction; 40 days with extraction |
| pH |
none required |
analyze immediately |
Other than adding a preservative, solution samples generally do not need additional preparation before analysis. This is the case for samples of natural waters and wastewaters. Solution samples with particularly complex matricies—blood and milk are two common examples—may need addi- tional processing to separate analytes from interferents, a topic covered later in this chapter.
Gases
Typical examples of gaseous samples include automobile exhaust, emissions from industrial smokestacks, atmospheric gases, and compressed gases. Also included in this category are aerosol particulates—the fine solid particles and liquid droplets that form smoke and smog. Let’s use the sampling of urban air as a case study in how to sample a gas.
Sample Collection
One approach for collecting a sample of urban air is to fill a stainless steel canister or a Tedlar/Teflon bag. A pump pulls the air into the container and, after purging, the container is sealed. This method has the advantage of being simple and of collecting a representative sample. Disadvantages include the tendency for some analytes to adsorb to the container’s walls, the presence of analytes at concentrations too low to detect with suitable accuracy and precision, and the presence of reactive analytes, such as ozone and nitrogen oxides, that may react with the container or that may otherwise alter the sample’s chemical composition during storage. When using a stainless steel canister, cryogenic cooling, which changes the sample from a gaseous state to a liquid state, may limit some of these disadvantages.
Most urban air samples are collected by filtration or by using a trap that contains a solid sorbent. Solid sorbents are used for volatile gases (a vapor pressure more than 10–6 atm) and for semi-volatile gases (a vapor pressure between 10–6 atm and 10–12 atm). Filtration is used to collect aerosol particulates. Trapping and filtering allow for sampling larger volumes of gas—an important concern for an analyte with a small concentration—and stabilizes the sample between its collection and its analysis.
In solid sorbent sampling, a pump pulls the urban air through a canister packed with sorbent particles. Typically 2–100 L of air are sampled when collecting a volatile compound and 2–500 m3 when collecting a semi-volatile gas. A variety of inorganic, organic polymer, and carbon sorbents have been used. Inorganic sorbents, such as silica gel, alumina, magnesium aluminum silicate, and molecular sieves, are efficient collectors for polar compounds. Their efficiency at absorbing water, however, limits their capacity for many organic analytes.
1 m3 is equivalent to 103 L.
Organic polymeric sorbents include polymeric resins of 2,4-diphenyl-p-phenylene oxide or styrene-divinylbenzene for volatile compounds, and polyurethane foam for semi-volatile compounds. These materials have a low affinity for water and are efficient for sampling all but the most highly volatile organic compounds and some lower molecular weight alcohols and ketones. Carbon sorbents are superior to organic polymer resins, which makes them useful for highly volatile organic compounds that will not absorb onto polymeric resins, although removing the compounds may be difficult.
Non-volatile compounds normally are present either as solid particulates or are bound to solid particulates. Samples are collected by pulling a large volume of urban air through a filtering unit and collecting the particulates on glass fiber filters.
The short term exposure of humans, animals, and plants to atmospheric pollutants is more severe than that for pollutants in other matrices. Because the composition of atmospheric gases can vary significantly over a time, the continuous monitoring of atmospheric gases such as O3, CO, SO2, NH3, H2O2, and NO2 by in situ sampling is important [Tanner, R. L. in Keith, L. H., ed. Principles of Environmental Sampling, American Chemical Society: Washington, D. C., 1988, 275–286].
Sample Preservation
After collecting a gross sample of urban air, generally there is little need for sample preservation or preparation. The chemical composition of a gas sample usually is stable when it is collected using a solid sorbent, a filter, or by cryogenic cooling. When using a solid sorbent, gaseous compounds are released for analysis by thermal desorption or by extracting with a suitable solvent. If the sorbent is selective for a single analyte, the increase in the sorbent’s mass is used to determine the amount of analyte in the sample.
Solids
Typical examples of solid samples include large particulates, such as those found in ores; smaller particulates, such as soils and sediments; tablets, pellets, and capsules used for dispensing pharmaceutical products and animal feeds; sheet materials, such as polymers and rolled metals; and tissue samples from biological specimens. Solids usually are heterogeneous and we must collect samples carefully if they are to be representative of the target population. Let’s use the sampling of sediments, soils, and ores as a case study in how to sample solids.
Sample Collection
Sediments from the bottom of streams, rivers, lakes, estuaries, and oceans are collected with a bottom grab sampler or with a corer. A bottom grab sampler (Figure \(\PageIndex{2}\)) is equipped with a pair of jaws that close when they contact the sediment, scooping up sediment in the process. Its principal advantages are ease of use and the ability to collect a large sample. Disadvantages include the tendency to lose finer grain sediment particles as water flows out of the sampler, and the loss of spatial information—both laterally and with depth—due to mixing of the sample.

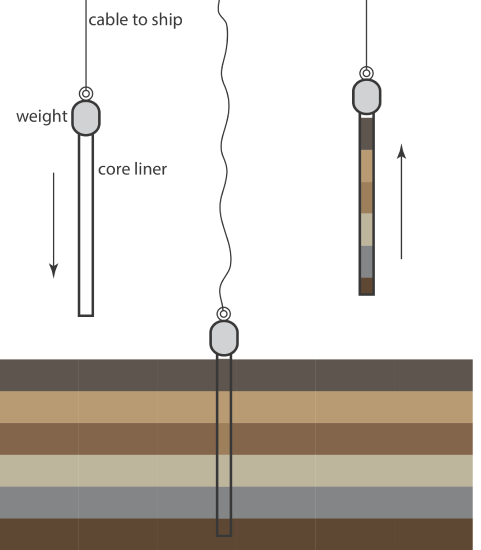
An alternative method for collecting sediments is the cylindrical coring device shown in Figure \(\PageIndex{3}\)). The corer is dropped into the sediment, collecting a column of sediment and the water in contact with the sediment. With the possible exception of sediment at the surface, which may experience mixing, samples collected with a corer maintain their vertical profile, which preserves information about how the sediment’s composition changes with depth.
Collecting soil samples at depths of up to 30 cm is accomplished with a scoop or a shovel, although the sampling variance generally is high. A better tool for collecting soil samples near the surface is a soil punch, which is a thin-walled steel tube that retains a core sample after it is pushed into the soil and removed. Soil samples from depths greater than 30 cm are collected by digging a trench and collecting lateral samples with a soil punch. Alternatively, an auger is used to drill a hole to the desired depth and the sample collected with a soil punch.
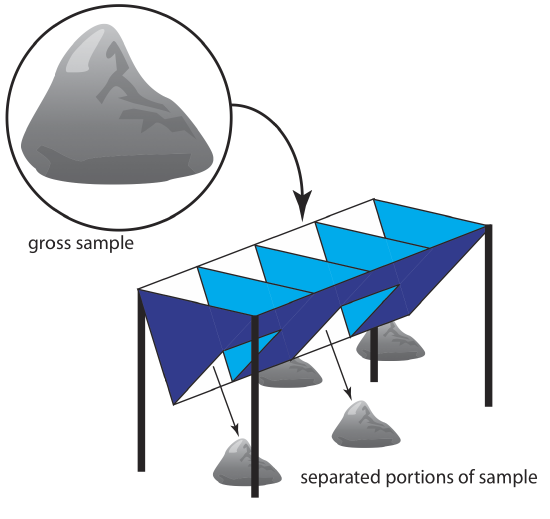
For particulate materials, particle size often determines the sampling method. Larger particulate solids, such as ores, are sampled using a riffle (Figure \(\PageIndex{4}\)), which is a trough with an even number of compartments. Because adjoining compartments empty onto opposite sides of the riffle, dumping a gross sample into the riffle divides it in half. By repeatedly passing half of the separated material back through the riffle, a sample of the desired size is collected.
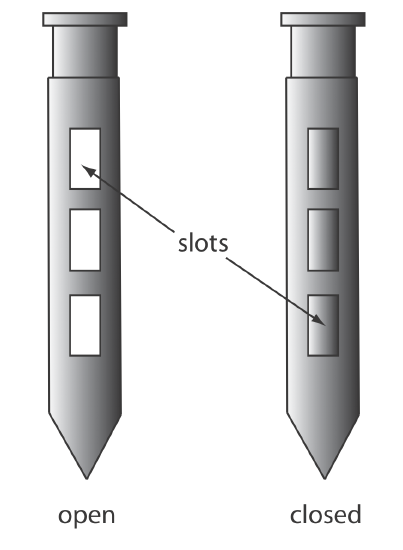
A sample thief (Figure \(\PageIndex{5}\)) is used for sampling smaller particulate materials, such as powders. A typical sample thief consists of two tubes that are nestled together. Each tube has one or more slots aligned down the length of the sample thief. Before inserting the sample thief into the material being sampled, the slots are closed by rotating the inner tube. When the sample thief is in place, rotating the inner tube opens the slots, which fill with individual samples. The inner tube is then rotated to the closed position and the sample thief withdrawn.
Sample Preservation
Without preservation, a solid sample may undergo a change in composition due to the loss of volatile material, biodegradation, or chemical reactivity (particularly redox reactions). Storing samples at lower temperatures makes them less prone to biodegradation and to the loss of volatile material, but fracturing of solids and phase separations may present problems. To minimize the loss of volatile compounds, the sample container is filled completely, eliminating a headspace where gases collect. Samples that have not been exposed to O2 particularly are susceptible to oxidation reactions. For example, samples of anaerobic sediments must be prevented from coming into contact with air.
Sample Preparation
Unlike gases and liquids, which generally require little sample preparation, a solid sample usually needs some processing before analysis. There are two reasons for this. First, as discussed in Chapter 7.2, the standard deviation for sampling, ssamp, is a function of the number of particles in the sample, not the combined mass of the particles. For a heterogeneous material that consists of large particulates, the gross sample may be too large to analyze. For example, a Ni-bearing ore with an average particle size of 5 mm may require a sample that weighs one ton to obtain a reasonable ssamp. Reducing the sample’s average particle size allows us to collect the same number of particles with a smaller, more manageable mass. Second, many analytical techniques require that the analyte be in solution.
Reducing Particle Size
A reduction in particle size is accomplished by crushing and grinding the gross sample. The resulting particulates are then thoroughly mixed and divided into subsamples of smaller mass. This process seldom occurs in a single step. Instead, subsamples are cycled through the process several times until a final laboratory sample is obtained.
Crushing and grinding uses mechanical force to break larger particles into smaller particles. A variety of tools are used depending on the particle’s size and hardness. Large particles are crushed using jaw crushers that can reduce particles to diameters of a few millimeters. Ball mills, disk mills, and mortars and pestles are used to further reduce particle size.
A significant change in the gross sample’s composition may occur during crushing and grinding. Decreasing particle size increases the available surface area, which increases the risk of losing volatile components. This problem is made worse by the frictional heat that accompanies crushing and grinding. Increasing the surface area also exposes interior portions of the sample to the atmosphere where oxidation may alter the gross sample’s composition. Other problems include contamination from the materials used to crush and grind the sample, and differences in the ease with which particles are reduced in size. For example, softer particles are easier to reduce in size and may be lost as dust before the remaining sample is processed. This is a particular problem if the analyte’s distribution between different types of particles is not uniform.
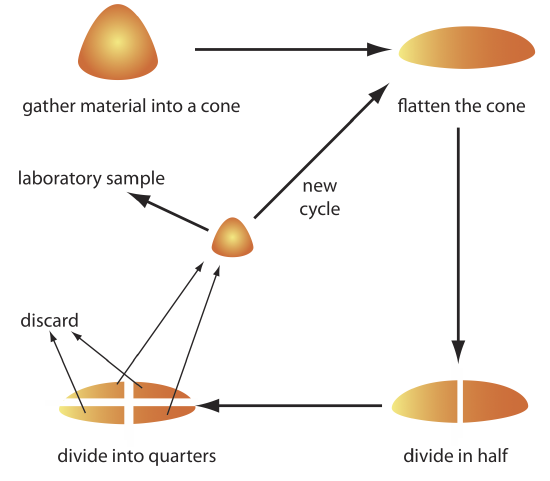
The gross sample is reduced to a uniform particle size by intermittently passing it through a sieve. Those particles not passing through the sieve receive additional processing until the entire sample is of uniform size. The resulting material is mixed thoroughly to ensure homogeneity and a subsample obtained with a riffle, or by coning and quartering. As shown in Figure \(\PageIndex{6}\), the gross sample is piled into a cone, flattened, and divided into four quarters. After discarding two diagonally opposed quarters, the remaining material is cycled through the process of coning and quartering until a suitable laboratory sample remains.
Bringing Solid Samples Into Solution
If you are fortunate, your sample will dissolve easily in a suitable solvent, requiring no more effort than gently swirling and heating. Distilled water usually is the solvent of choice for inorganic salts, but organic solvents, such as methanol, chloroform, and toluene, are useful for organic materials.
When a sample is difficult to dissolve, the next step is to try digesting it with an acid or a base. Table \(\PageIndex{2}\) lists several common acids and bases, and summarizes their use. Digestions are carried out in an open container, usually a beaker, using a hot-plate as a source of heat. The main advantage of an open-vessel digestion is cost because it requires no special equipment. Volatile reaction products, however, are lost, which results in a determinate error if they include the analyte.
| solution | uses and properties |
|---|---|
|
HCl (37% w/w) |
|
|
HNO3 (70% w/w) |
|
|
H2SO4 (98% w/w) |
|
| HF (50% w/w) |
|
|
HClO4 (70% w/w) |
|
|
HCl:HNO3 (3:1 v/v) |
|
|
NaOH |
|
Many digestions now are carried out in a closed container using microwave radiation as the source of energy. Vessels for microwave digestion are manufactured using Teflon (or some other fluoropolymer) or fused silica. Both materials are thermally stable, chemically resistant, transparent to microwave radiation, and capable of withstanding elevated pressures. A typical microwave digestion vessel, as shown in Figure \(\PageIndex{7}\), consists of an insulated vessel body and a cap with a pressure relief valve. The vessels are placed in a microwave oven (a typical oven can accommodate 6–14 vessels) and microwave energy is controlled by monitoring the temperature or pressure within one of the vessels.
 |
 |
Figure \(\PageIndex{7}\). Microwave digestion unit: on the left is a view of the unit’s interior showing the carousel that holds the digestion vessels; on the right is a close-up of a Teflon digestion vessel, which is encased in a thermal sleeve. The pressure relief value, which is part of the vessel’s blue cap, contains a membrane that ruptures if the internal pressure becomes too high.
Inorganic samples that resist decomposition by digesting with acids or bases often are brought into solution by fusing with a large excess of an alkali metal salt, called a flux. After mixing the sample and the flux in a crucible, they are heated to a molten state and allowed to cool slowly to room temperature. The resulting melt usually dissolves readily in distilled water or dilute acid. Table \(\PageIndex{3}\) summarizes several common fluxes and their uses. Fusion works when other methods of decomposition do not because of the high temperature and the flux’s high concentration in the molten liquid. Disadvantages include contamination from the flux and the crucible, and the loss of volatile materials.
| flux | melting temperature (oC) | crucible | typical samples |
|---|---|---|---|
| Na2CO3 | 85 | Pt | silicates, oxides, phosphate, sulfides |
| Li2B4O7 | 930 | Pt, graphite | aluminosilicates, carbontes |
| LiBO2 | 845 | Pt, graphite | aluminosilicates, carbonates |
| NaOH | 318 | Au, Ag | silicates, silicon carbide |
| KOH | 380 | Au, Ag | silicates, silicon carbide |
| Na2O2 | — | Ni | silicates, chromium steels, Pt alloys |
| K2S2O7 | 300 | Ni, porcelain | oxides |
| B2O3 | 577 | Pt | silicates, oxides |
Finally, we can decompose organic materials by dry ashing. In this method the sample is placed in a suitable crucible and heated over a flame or in a furnace. The carbon present in the sample oxidizes to CO2, and hydrogen, sulfur, and nitrogen are volatilized as H2O, SO2, and N2. These gases can be trapped and weighed to determine their concentration in the organic material. Often the goal of dry ashing is to remove the organic material, leaving behind an inorganic residue, or ash, that can be further analyzed.