
26.3 Synthesis of Amino Acids
- Page ID
- 91058
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Objectives
After completing this section, you should be able to
- outline, by means of equations, how a racemic mixture of given amino acid can be prepared from a carboxylic acid using reactions you studied earlier in the course.
-
- outline, by means of equations, the preparation of a given amino acid by the amidomalonate synthesis.
- identify the amino acid formed from using a given alkyl halide in an amidomalonate synthesis.
- identify the alkyl halide needed to produce a given amino acid by the amidomalonate synthesis.
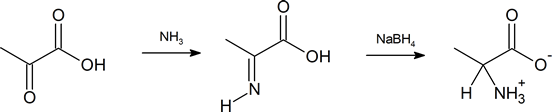
- describe, by means of equations, how an α‑keto acid can be transformed to an amino acid by reductive amination.
-
- describe a general method for resolving a racemic mixture of a given amino acid.
- provide a brief example of how a biological method may be employed to resolve a racemic mixture of a given amino acid.
- show the enantioselective preparation of an amino acid from the corresponding Z enamido acid.
Make certain that you can define, and use in context, the key terms below.
- amidomalonate synthesis
- enantioselective synthesis
- racemic mixture
Do not be alarmed by the number of methods to synthesize amino acids described in this section.You have seen many of these reactions in previous sections and should already be familiar with the approaches discussed here.
To fulfill the requirements of Objective 1, review the Hell‑Volhard‑Zelinskii reaction (Section 22.4) and the Gabriel phthalimide synthesis (Section 24.6).
The amidomalonate synthesis is a simple variation of the malonic ester synthesis (Section 22.7). A base abstracts a proton from the alpha carbon, which is then alkylated with an alkyl halide. Then both the hydrolysis of the esters and the amide protecting group under aqueous acidic conditions generates the α‑amino acid.
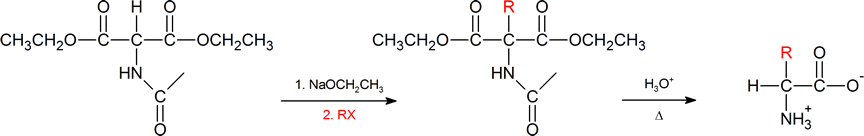
Another method of getting to the α‑amino acid is by reductive amination of the α‑keto acid which you have also previously encountered (Section 24.6).
Synthesis of α-Amino Acids
1) Amination of alpha-bromocarboxylic acids, illustrated by the following equation, provides a straightforward method for preparing alpha-aminocarboxylic acids. The bromoacids, in turn, are conveniently prepared from carboxylic acids by reaction with Br2 + PCl3. Although this direct approach gave mediocre results when used to prepare simple amines from alkyl halides, it is more effective for making amino acids, thanks to the reduced nucleophilicity of the nitrogen atom in the product. Nevertheless, more complex procedures that give good yields of pure compounds are often chosen for amino acid synthesis.
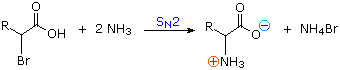
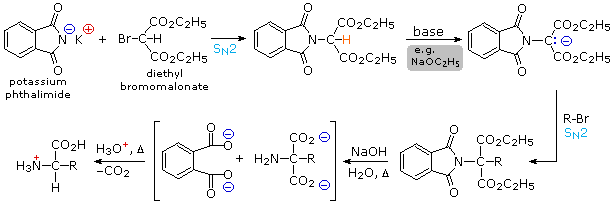
2) By modifying the nitrogen as a phthalimide salt, the propensity of amines to undergo multiple substitutions is removed, and a single clean substitution reaction of 1º- and many 2º-alkylhalides takes place. This procedure, known as the Gabriel synthesis, can be used to advantage in aminating bromomalonic esters, as shown in the upper equation of the following scheme. Since the phthalimide substituted malonic ester has an acidic hydrogen (colored orange), activated by the two ester groups, this intermediate may be converted to an ambident anion and alkylated. Finally, base catalyzed hydrolysis of the phthalimide moiety and the esters, followed by acidification and thermal decarboxylation, produces an amino acid and phthalic acid (not shown).
3) An elegant procedure, known as the Strecker synthesis, assembles an alpha-amino acid from ammonia (the amine precursor), cyanide (the carboxyl precursor), and an aldehyde. This reaction (shown below) is essentially an imino analog of cyanohydrin formation. The alpha-amino nitrile formed in this way can then be hydrolyzed to an amino acid by either acid or base catalysis.
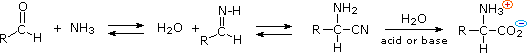
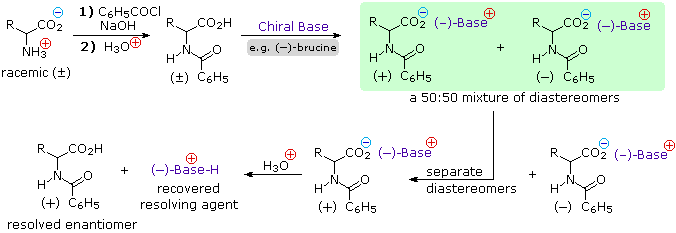
4) Resolution The three synthetic procedures described above, and many others that can be conceived, give racemic amino acid products. If pure L or D enantiomers are desired, it is necessary to resolve these racemic mixtures. A common method of resolving racemates is by diastereomeric salt formation with a pure chiral acid or base. This is illustrated for a generic amino acid in the following diagram. Be careful to distinguish charge symbols, shown in colored circles, from optical rotation signs, shown in parenthesis.
In the initial display, the carboxylic acid function contributes to diastereomeric salt formation. The racemic amino acid is first converted to a benzamide derivative to remove the basic character of the amino group. Next, an ammonium salt is formed by combining the carboxylic acid with an optically pure amine, such as brucine (a relative of strychnine). The structure of this amine is not shown, because it is not a critical factor in the logical progression of steps. Since the amino acid moiety is racemic and the base is a single enantiomer (levorotatory in this example), an equimolar mixture of diastereomeric salts is formed (drawn in the green shaded box). Diastereomers may be separated by crystallization, chromatography or other physical manipulation, and in this way one of the isomers may be isolated for further treatment, in this illustration it is the (+):(-) diastereomer. Finally the salt is broken by acid treatment, giving the resolved (+)-amino acid derivative together with the recovered resolving agent (the optically active amine). Of course, the same procedure could be used to obtain the (-)-enantiomer of the amino acid.
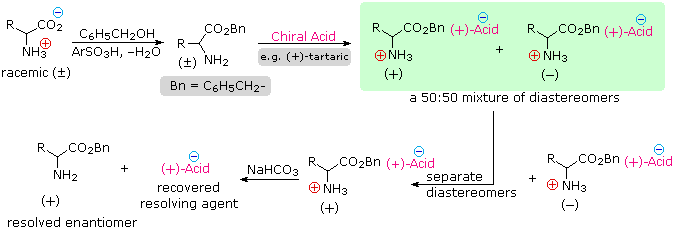
Since amino acids are amphoteric, resolution could also be achieved by using the basic character of the amine function. For this approach we would need an enantiomerically pure chiral acid such as tartaric acid to use as the resolving agent. This alternative resolution strategy will be illustrated. Note that the carboxylic acid function is first esterified, so that it will not compete with the resolving acid.
Resolution of aminoacid derivatives may also be achieved by enzymatic discrimination in the hydrolysis of amides. For example, an aminoacylase enzyme from pig kidneys cleaves an amide derivative of a natural L-amino acid much faster than it does the D-enantiomer. If the racemic mixture of amides shown in the green shaded box above is treated with this enzyme, the L-enantiomer (whatever its rotation) will be rapidly converted to its free zwitterionic form, whereas the D-enantiomer will remain largely unchanged. Here, the diastereomeric species are transition states rather than isolable intermediates. This separation of enantiomers, based on very different rates of reaction, is called kinetic resolution.
Enantioselective Synthesis
Till now all of the synthetic routes to α-amino acids we have discussed yield a racemic mixture. Once produced one could resolve the mixture to obtain pure L or D enantiomers. However, enantioselective synthetic methods to produce pure compounds directly are being developed. For instance, several catalysts are now available for reduction of C=C to expose enantiopure amino acids. A good example is the industrial synthesis of L-DOPA, a drug used in the treatment of Parkinson’s disease. W.S. Knowles shared the 2001 Nobel Price with R. Noyori and K.B. Sharpless for their contributions in the area of asymmetric catalytic reductions. Knowles developed several chiral phosphine–metal catalysts for asymmetric reductions. The rhodium(I) catalyst shown, which is complexed by large organic ligands, facilitates production of almost pure L-DOPA.
Contributors and Attributions
Dr. Dietmar Kennepohl FCIC (Professor of Chemistry, Athabasca University)
Prof. Steven Farmer (Sonoma State University)
William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry