
21.7: The Microscopic Model for Reversible Change
- Page ID
- 152781

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Now let us return to the closed (constant-\(N\)) system to develop another perspective on the dependence of its macroscopic thermodynamic properties on the molecular energy levels and their probabilities. We undertake to describe the system using volume and temperature as the independent variables. In thinking about the energy-level probabilities, we stipulate that any parameters that affect the state of the system remain constant. Specifically, we mean that any parameters that appear in the Schrödinger equation remain constant. For example, the energy levels of a particle in a box depend on the mass of the particle and the length of the box. Any such parameter is called an exogenous variable. If we change an exogenous variable (say the length of the box) by a small amount, all of the energy levels change by a small amount, and all of the probabilities change by a small amount. The energy levels and their probabilities are smooth functions of the exogenous variable. If \(\xi\) is the exogenous variable, we have
\[P_i=P\left(\epsilon_i\right)=g_i\rho \left(\epsilon_i\left(\xi \right)\right) \nonumber \]
A change in the exogenous variable corresponds to a reversible macroscopic process.
For a particle in a box, the successive \({\psi }_i\) are functions that depend on the quantum number, \(i\), and the length of the box, \(\ell\). When we change the length of the box, the wavefunction and its associated energy both change. Both are continuous functions of the length of the box. The energy is
\[\epsilon_i=\frac{i^2h^2}{8m{\ell }^2} \nonumber \]
Changing the length of the box is analogous to changing the volume of a system. A reversible volume change entails work. We see that changing the length of the box does work on the particle-in-a-box, just as changing the volume of a three-dimensional system does work on the system.
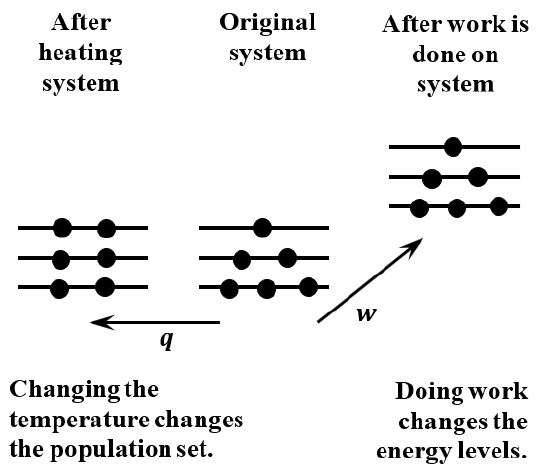
Temperature plays a central role in the description of equilibrium from the macroscopic perspective. We can see that temperature enters the description of equilibrium from the microscopic perspective through its effect on the probability factors. When we increase the temperature of a system, its energy increases. The average energy of its molecules increases. The probability of an energy level must depend on temperature. Evidently, the probabilities of energy levels that are higher than the original average energy increase when the temperature increases. The probabilities of energy levels that are lower than the original average energy decrease when the temperature increases. The effects of heat and work on the energy levels and their equilibrium populations are diagrammed in Figure \(\PageIndex{1}\).
If our theory is to be useful, the energy we measure for a macroscopic system must be indistinguishably close to the expected value of the system energy as calculated from our microscopic model:
\[E_{\mathrm{experiment}}\approx \left\langle E\right\rangle =N\left\langle \epsilon \right\rangle =N\sum^{\infty }_{i=1}{P_i\epsilon_i} \nonumber \]
We can use this equation to relate the probabilities, \(P_i\), to other thermodynamic functions. Dropping the distinction between the experimental and expected energies, and assuming that the \(\epsilon_i\) and the \(P_i\) are continuous variables, we find the total differential
\[dE=N\sum^{\infty }_{i=1}{\epsilon_idP_i}+N\sum^{\infty }_{i=1}{{P_id\epsilon }_i} \nonumber \]
This equation is important because it describes a reversible macroscopic process in terms of the microscopic variables \(\epsilon_i\) and \(P_i\).
Let us consider the first term. Since \(N\) is a constant, we have from \(N^{\textrm{⦁}}_i=P_iN\) that \(dN^{\textrm{⦁}}_i=NdP_i\). Substituting, we have
\[\left(dE\right)_{\epsilon_i}=N\sum^{\infty }_{i=1}{\epsilon_idP_i}=\sum^{\infty }_{i=1}{\epsilon_i}dN^{\textrm{⦁}}_i \nonumber \]
This asserts that the energy of the system changes if we redistribute the molecules among the various energy levels. If the redistribution takes molecules out of lower energy levels and puts them into higher energy levels, the energy of the system increases. This is our statistical-mechanical picture of the shift in the equilibrium position that occurs when we heat a system of independent molecules; the allocation of molecules among the available energy levels shifts to put more molecules in higher energy levels and fewer in lower ones. This corresponds to an increase in the temperature of the macroscopic system.
In terms of the macroscopic system, the first term represents an increment of heat added to the system in a reversible process; that is,
\[dq^{rev}=N\sum^{\infty }_{i=1}{\epsilon_idP_i} \nonumber \]
The second term, \(N\sum^{\infty }_{i=1}{{P_id\epsilon }_i}\), is a contribution to the change in the energy of the system from reversible changes in the energy of the various quantum states, while the number of molecules in each quantum state remains constant. This term corresponds to a process in which the quantum states (and their energies) evolve in a continuous way as the state of the system changes. The second term represents an increment of work done on the system in a reversible process; that is
\[dw^{rev}=N\sum^{\infty }_{i=1}{{P_id\epsilon }_i} \nonumber \]
Evidently, the total differential expression for \(dE\) is the fundamental equation of thermodynamics expressed in terms of the variables we use to characterize the molecular system. It enables us to relate the variables that characterize our microscopic model of the molecular system to the variables that characterize the macroscopic system.
For a system in which the reversible work is pressure–volume work, the energy levels depend on the volume. At constant temperature we have
\[dw^{rev}=-PdV=N\sum^{\infty }_{i=1}{{P_id\epsilon }_i}=N\sum^{\infty }_{i=1}{P_i{\left(\frac{\partial \epsilon_i}{\partial V}\right)}_TdV} \nonumber \]
so that the system pressure, \(P\), is related to the energy-level probabilities, \(P_i\), as
\[P=-N\sum^{\infty }_{i=1}{P_i{\left(\frac{\partial \epsilon_i}{\partial V}\right)}_T} \nonumber \]
To evaluate the pressure, we must know how the energy levels depend on the volume of the system.
The first term relates the entropy to the energy-level probabilities. Since \(dq^{rev}=TdS=N\sum^{\infty }_{i=1}{\epsilon_idP_i}\), we have \[dS=\frac{N}{T}\sum^{\infty }_{i=1}{\epsilon_idP_i} \nonumber \]
From the Boltzmann distribution function we have
\(P_i=z^{-1}g_i\mathrm{exp}\left({-\epsilon_i}/{kT}\right)\), or
\[\epsilon_i=-kT\ln P_i +kT\ln g_i -kT\ln z \nonumber \]
Substituting into our expression for \(dS\), we find
\[dS=-Nk\sum^{\infty }_{i=1}{\left(\ln P_i \right)}dP_i+Nk\sum^{\infty }_{i=1}{\left(\ln g_i \right)}dP_i-Nk\left(\ln z \right)\sum^{\infty }_{i=1}{dP_i} \nonumber \]
Since \(\sum^{\infty }_{i=1}{P_i}=1\), we have \(\sum^{\infty }_{i=1}{dP_i}=0\), and the last term vanishes. Also,
\[\sum^{\infty }_{i=1}{d\left(P_i\ln P_i \right)}=\sum^{\infty }_{i=1}{\left(\ln P_i \right){dP}_i}+\sum^{\infty }_{i=1}{dP_i}=\sum^{\infty }_{i=1}{\left(\ln P_i \right){dP}_i} \nonumber \]
so that
\[dS=-Nk\sum^{\infty }_{i=1}{d\left(P_i\ln P_i \right)}+Nk\sum^{\infty }_{i=1}{\left(\ln g_i \right)}dP_i \nonumber \]
At any temperature, the probability ratio for any two successive energy levels is
\[\frac{P_{i+1}\left(T\right)}{P_i\left(T\right)}=\frac{P_{i+1}}{P_i}=\frac{g_{i+1}}{g_i}\mathrm{exp}\left(\frac{-\left(\epsilon_{i+1}-\epsilon_i\right)}{kT}\right) \nonumber \]
In the limit as the temperature goes to zero,
\[\frac{P_{i+1}}{P_i}\to 0 \nonumber \]
It follows that \(P_1\left(0\right)=1\) and \(P_i\left(0\right)=0\) for \(i>1\). Integrating from \(T=0\) to \(T\), the entropy of the system goes from \(S\left(0\right)=S_0\) to \(S\left(T\right)\), and the energy-level probabilities go from \(P_i\left(0\right)\) to \(P_i\left(T\right)\). We have
\[\int^{S\left(T\right)}_{S_0}{dS}=-Nk\sum^{\infty }_{i=1}{\int^{P_i\left(T\right)}_{P_i\left(0\right)}{d\left(P_i\ln P_i \right)}}+Nk\sum^{\infty }_{i=1}{\int^{P_i\left(T\right)}_{P_i\left(0\right)}{\left(\ln g_i \right)dP_i}} \nonumber \]
so that
\[S\left(T\right)-S_0=-Nk\sum^{\infty }_{i=1}{P_i\left(T\right)}\ln P_i\left(T\right) +NkP_1\left(0\right)\ln P_1\left(0\right) +Nk\sum^{\infty }_{i=1}{\left(\ln g_i \right)P_i\left(T\right)}-Nk\left(\ln g_1 \right)P_1\left(0\right) \nonumber \]
Since \(P_1\left(0\right)=1\), \(\ln P_1\left(0\right) \) vanishes. The entropy change becomes
\[S\left(T\right)-S_0=-Nk\sum^{\infty }_{i=1}{P_i}\left[\ln P_i -\ln g_i \right]-Nk\ln g_1 =-Nk\sum^{\infty }_{i=1}{P_i}\ln \rho \left(\epsilon_i\right) -Nk\ln g_1 \nonumber \]
We have \(S_0=Nk\ln g_1 \). If \(g_1=1\), the lowest energy level is non-degenerate, and \(S_0=0\); then we have
\[S=-Nk\sum^{\infty }_{i=1}{P_i}\ln \rho \left(\epsilon_i\right) \nonumber \]
This is the entropy of an \(N\)-molecule, constant-volume, constant-temperature system that is in thermal contact with its surroundings at the same temperature. We obtain this same result in Sections 20.10 and 20.14 by arguments in which we assume that the system is isolated. In all of these arguments, we assume that the constant-temperature system and its isolated counterpart are functionally equivalent; that is, a group of population sets that accounts for nearly all of the probability in one system also accounts for nearly all of the probability in the other.
Because we obtain this result by assuming that the system is composed of \(N\), independent, non-interacting, distinguishable molecules, the entropy of this is system is \(N\) times the entropy contribution of an individual molecule. We can write
\[S_{\mathrm{molecule}}=-k\sum^{\infty }_{i=1}{P_i}\ln \rho \left(\epsilon_i\right) \nonumber \]