
20.1: The Independent-Molecule Approximation
- Page ID
- 151788

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)In Chapter 18, our survey of quantum mechanics introduces the idea that a molecule can have any of an infinite number of discrete energies, which we can put in order starting with the smallest. We now turn our attention to the properties of a system composed of a large number of molecules. This multi-molecule system must obey the laws of quantum mechanics. Therefore, there exists a Schrödinger equation, whose variables include all of the inter-nucleus, inter-electron, and electron-nucleus distance and potential terms in the entire multi-molecule system. The relevant boundary conditions apply at the physical boundaries of the macroscopic system. The solutions of this equation include a set of infinitely many wavefunctions, \({\mathit{\Psi}}_{i,j}\), each describing a quantum mechanical state of the entire multi-molecule system. In general, the collection of elementary particles that can be assembled into a particular multi-molecule system can also be assembled into many other multi-molecule systems. For example, an equimolar mixture of \(CO\) and \(H_2O\) can be reassembled into a system comprised of equimolar \(CO_2\) and \(H_2\), or into many other systems containing mixtures of \(CO\), \(H_2O\), \(CO_2\), and \(H_2\). Infinitely many quantum-mechanical states are available to each of these multi-molecule systems.
For every such multi-molecule wavefunction, \({\psi }_{i,j}\), there is a corresponding system energy, \(E_i\). In general, the system energy, \(E_i\), is \({\mathit{\Omega}}_i\)-fold degenerate; there are \({\mathit{\Omega}}_i\) wavefunctions, \({\mathit{\Psi}}_{i,1}\), \({\mathit{\Psi}}_{i,2}\), …, \({\mathit{\Psi}}_{i,{\mathit{\Omega}}_i}\), whose energy is \(E_i\). The wavefunctions include all of the interactions among the molecules of the system, and the energy levels of the system reflect all of these interactions. While generating and solving this multi-molecule Schrödinger equation is straightforward in principle, it is completely impossible in practice.
Fortunately, we can model multi-molecule systems in another way. The primary focus of chemistry is the study of the properties and reactions of molecules. Indeed, the science of chemistry exists, as we know it, only because the atoms comprising a molecule stick together more tenaciously than molecules stick to one another. (Where this is not true, we get macromolecular materials like metals, crystalline salts, etc.) This occurs because the energies that characterize the interactions of atoms within a molecule are much greater than the energies that characterize the interaction of one molecule with another. Consequently, the energy of the system can be viewed as the sum of two terms. One term is a sum of the energies that the component molecules would have if they were all infinitely far apart. The other term is a sum of the energies of all of the intermolecular interactions, which is the energy change that would occur if the molecules were brought from a state of infinite separation to the state of interest.
In principle, we can describe a multi-molecule system in this way with complete accuracy. This description has the advantage that it breaks a very large and complex problem into two smaller problems, one of which we have already solved: In Chapter 18, we see that we can approximate the quantum-mechanical description of a molecule and its energy levels by factoring molecular motions into translational, rotational, vibrational, and electronic components. It remains only to describe the intermolecular interactions. When intramolecular energies are much greater than intermolecular-interaction energies, it may be a good approximation to ignore the intermolecular interactions altogether. This occurs when we describe ideal gas molecules; in the limit that a gas behaves ideally, the force between any two of its molecules is nil.
In Chapter 23, we return to the idea of multi-molecule wavefunctions and energy levels. Meanwhile we assume that intermolecular interactions can be ignored. This is a poor approximation for many systems. However, it is a good approximation for many others, and it enables us to keep our description of the system simple while we use molecular properties in our development of the essential ideas of statistical thermodynamics.
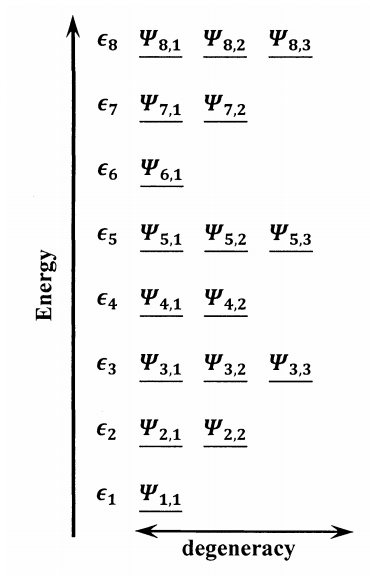
We focus on developing a theory that gives the macroscopic thermodynamic properties of a pure substance in terms of the energy levels available to its individual molecules. To begin, we suppose that we solve the Schrödinger equation for an isolated molecule. In this Schrödinger equation, the variables include the inter-nucleus, inter-electron, and electron-nucleus distance and potential terms that are necessary to describe the molecule. The solutions are a set of infinitely many wavefunctions, \({\psi }_{i,j}\), each describing a different quantum-mechanical state of an isolated molecule. We refer to each of the possible wavefunctions as quantum state of the molecule. For every such wavefunction, there is a corresponding molecular energy, \({\epsilon }_i\). Every unique molecular energy, \({\epsilon }_i\), is called an energy level. Several quantum states can have the same energy. When two or more quantum states have the same energy, we say that they belong to the same energy level, and the energy level is said to be degenerate. In general, there are \(g_i\) quantum states that we can represent by the \(g_i\) wavefunctions, \({\psi }_{i,1}\), , \({\psi }_{i,2}\), ..., , \({\psi }_{i,g_i}\), each of whose energy is \({\epsilon }_i\). The number of quantum states that have the same energy is called the degeneracy of the energy level. Figure 1 illustrates the terms we use to describe the quantum states and energy levels available to a molecule.
In our development of classical thermodynamics, we find it convenient to express the value of a thermodynamic property of a pure substance as the change that occurs during a formal process that forms one mole of the substance, in its standard state, from its unmixed constituent elements, in their standard states. In developing statistical thermodynamics, we find it convenient to express the value of a molecular energy, \({\epsilon }_i\), as the change that occurs during a formal process that forms a molecule of the substance, in one of its quantum states, \({\psi }_{i,j}\), from its infinitely separated, stationary, constituent atoms. That is, we let the isolated constituent atoms be the reference state for the thermodynamic properties of a pure substance.