
29.2: Vibrational Spectroscopy
- Page ID
- 416126

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
Vibrational spectroscopy is concerned with the measurement of the energies of transitions between quantized vibrational states of molecules in the gas phase. These transitions usually occur in the middle infrared (IR) region of the electromagnetic wave at approximately \(4,000-400\;\text{cm}^{-1}\) (\(2.5-25\;\mu \text{m}\)). In the gas phase, vibrational transitions are almost always accompanied by changes in rotational energy. Transitions involving changes in both vibrational and rotational states are usually abbreviated as rovibrational transitions. Since changes in rotational energy levels are typically much smaller than changes in vibrational energy levels, changes in rotational state are said to give fine structure to the vibrational spectrum. For a given vibrational transition, the same theoretical treatment that we saw in the previous section for pure rotational spectroscopy gives the rotational quantum numbers, energy levels, and selection rules.
As we have done in the previous section, we will discuss below the simplest case of a diatomic molecule. For non-linear molecules the spectra becomes complicated to calculate, but their interpretation remains an important tool for the analysis of chemical structures.
Vibration of heteronuclear diatomic molecules
Diatomic molecules with the general formula \(\mathrm{AB}\) have one normal mode of vibration involving stretching of the \(\mathrm{A}-\mathrm{B}\) bond. The vibrational term values, \(G(v)\) can be calculated with the harmonic approximation that we discussed in chapter 20. The resulting equidistant energy levels depend on one vibrational quantum number \(v\):
\[ G(v) = \omega_e \left( v + \dfrac{1}{2} \right), \label{30.2.1} \]
where \(\omega_e\) is the harmonic frequency around equilibrium. When the molecule is in the gas phase, it can rotate about an axis, perpendicular to the molecular axis, passing through the center of mass of the molecule. As we discussed in the previous section, the rotational energy is also quantized, and depend on the rotational quantum number \(J\). The values of the ro-vibrational states are found (in wavenumbers) by combining the expressions for vibration and rotation:
\[ G(v)+F_{v}(J)=\left[\omega_e \left(v + \dfrac{1}{2} \right) +B_{v}J(J+1)\right], \label{30.2.2} \]
where \(F_{v}(J)\) are the rotational levels at each vibrational state \(v\).\(^1\)
The selection rule for electric dipole allowed ro-vibrational transitions, in the case of a diamagnetic diatomic molecule is:
\[ \Delta v=\pm 1\ (\pm 2,\pm 3,\ldots),\; \Delta J=\pm 1. \label{30.2.3} \]
The transition with \(\Delta v =\pm 1\) is known as the fundamental transition, while the others are called overtones. The selection rule has two consequences:
- Both the vibrational and rotational quantum numbers must change. The transition \(\Delta v=\pm 1,\;\Delta J=0\) (Q-branch) is forbidden.
- The energy change of rotation can be either subtracted from or added to the energy change of vibration, giving the P- and R- branches of the spectrum, respectively.
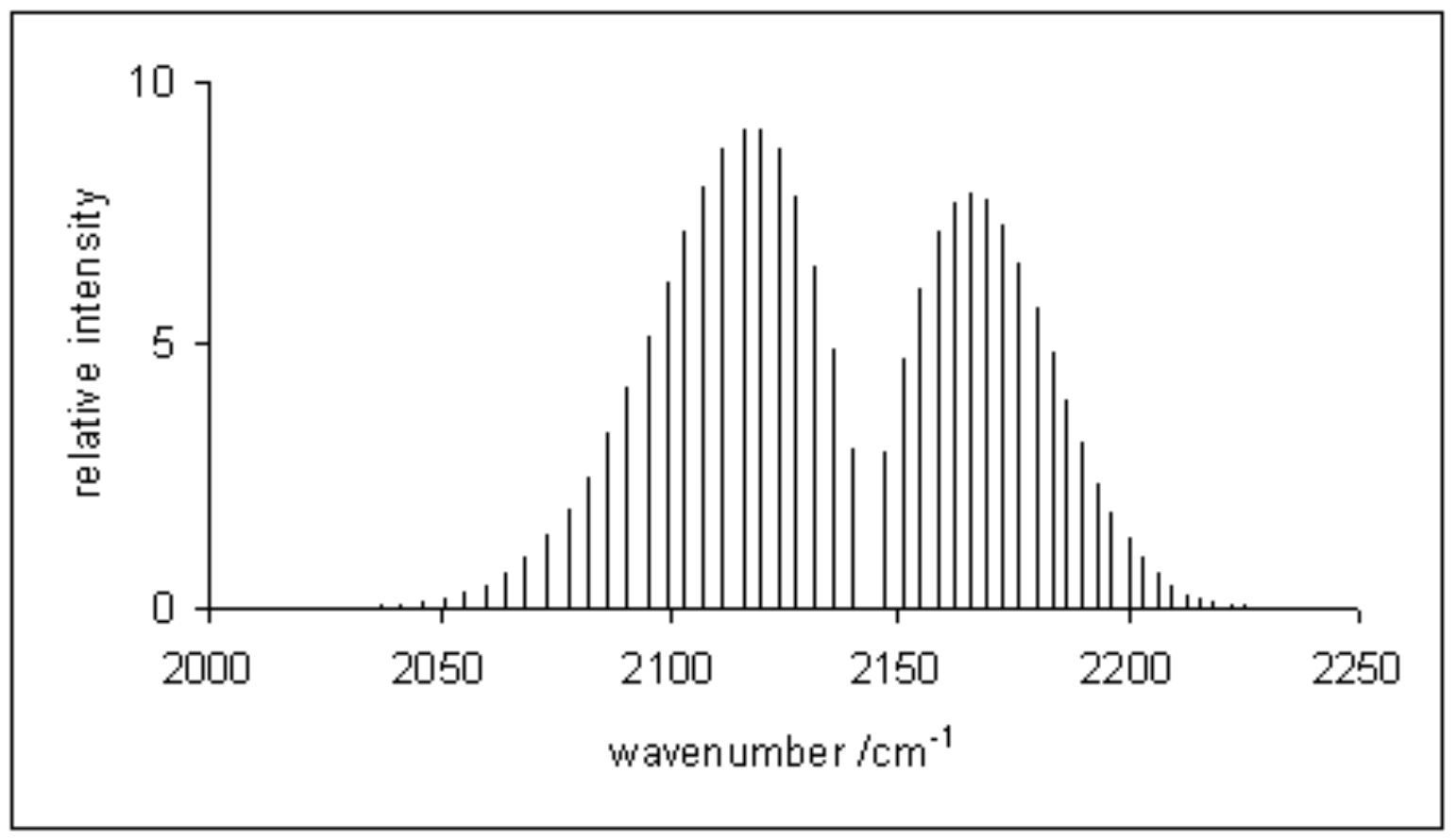
A typical rovibrational spectrum is reported in figure \(\PageIndex{1}\) for the \(\mathrm{CO}\) molecule.\(^2\) The intensity of the signals is—once again—proportional to the initial population of the levels. Notice how the signals in the spectrum are divided among two sides, the P-branch to the left, and the R-branch to the right. These signals correspond to the transitions reported in figure \(\PageIndex{2}\).\(^3\) Notice how the transitions corresponding to the Q-branch are forbidden by the selection rules, and therefore not observed in the experimental spectrum. The position of the missing Q-branch, however, can be easily obtained from the experimental spectrum as the missing signal between the P- and R- branches. Since the Q-branch transitions do not involve changes in the rotational energy level, their value is directly proportional to \(\omega_e\). This fact makes rovibrational spectroscopy an important experimental tool in the determination of bond distances of diatomic molecules.

Vibration of homonuclear diatomic molecules
The quantum mechanics for homonuclear diatomic molecules is qualitatively the same as for heteronuclear diatomic molecules, but the selection rules governing transitions are different. Since the electric dipole moment of the homonuclear diatomics is zero, the fundamental vibrational transition is electric-dipole-forbidden and the molecules are infrared inactive.
The spectra of these molecules can be observed by a type of IR spectroscopy that is subject to different selection rules. This technique is called Raman spectroscopy, and allows identification of the rovibrational spectra of homonuclear diatomic molecules because their molecular vibration is Raman-allowed.
- ︎This is just a first approximation to rovibrational spectroscopy. Corrections for anharmonicity centrifugal distortion are necessary to closely match experimental spectra.︎
- This picture is taken from Wikipedia of anonimous user, and distributed under CC BY 3.0 license.︎
- This picture is taken from Wikipedia by user David-i98, and under public domain.