
8. Phase Transitions
- Page ID
- 8667
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)When chemical reactions occur, the system makes transition among multiple minima at the molecular level. The figure below illustrates the molecular connection between free energy \(G\), its derivative \(\Delta G\), and the free energy as a function of reaction coordinate, \(G(x)\).
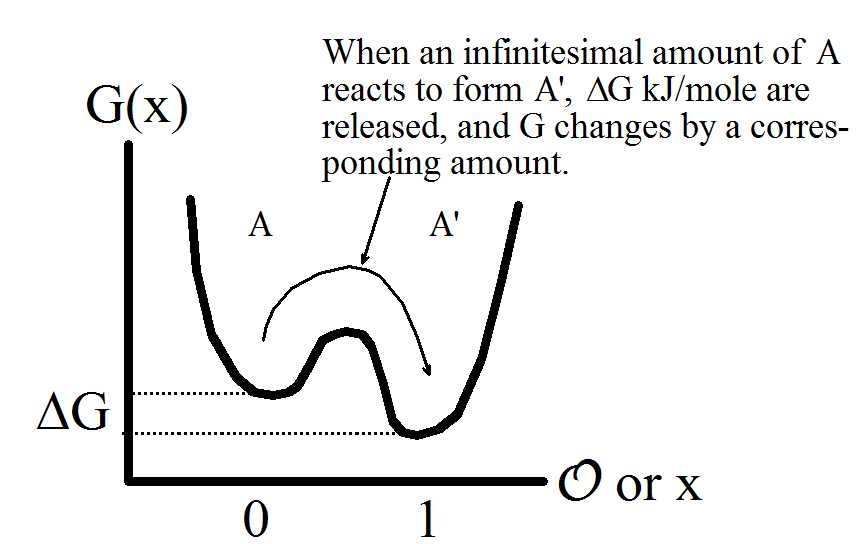
Figure 8.1: The free energy \(G(x)\) as a function of coordinate (e.g. bond distance) has two local minima, A’ being lower in free energy. When A converts to A’, \(\Delta G\) kJ/mole are released per infinitesimal amount of reaction \(d\xi\). Note that both x and \(\xi\) can vary between 0 and 1, but the meaning is very different: when all of the substance is in state A, \(x=\xi=0\), and when it is all in state A', \(x=\xi=1\). However, while the substance can have \(\xi=0.5\) when the reaction is half completed, almost none of it will ever be at x=0.5. Rather, half will be at x=1 and half at x=1.
So far, we have treated macroscopic pure substances and mixtures as though they had a single minimum in the free energy as a function of reaction coordinate. However, thermodynamics does not forbid multiple minima even at the macroscopic level, and can be used to make comparative statements about the minima.
| Definition: Phase |
|---|
|
A phase is a local minimum in the free energy surface. |
Unlike ordinary chemical reactions, transition between phases can occur even when only one pure substance is present:
\[ A^{(1)} \rightarrow A^{(2)} \]
For phase transitions, we call the reaction coordinate “order parameter.” The superscripts refer to the phases.
| Definition: Order Parameter |
|---|
|
An order parameter is a thermodynamic variable scaled to zero in one phase, nonzero in an(other) phase. |
| Example 8.1 |
|---|
|
Example: a gas-liquid transition order parameter \[ O = \rho - \rho_{gas}\] or \[ O = \dfrac{ \rho - \rho_{gas}}{ \rho_{liq} - \rho_{gas}}\] in general \[ O = X - X^{(2)}\] or \[ O = \dfrac{ X - X^{(2)}}{ X^{(2)} - X^{(1)}}\] Thermodynamics cannot make statements about the details of the barrier (e.g. its height), or how fast a transition can occur. The transition itself is a rather delicate matter – it violates P2 since temporarily \(\Delta G >0\) if the transition occurs at constant \(T\) and \(P\). The solution to this dilemma: if climbing the barrier were required of the entire macroscopic system, phase transitions could indeed never occur. Rather, a small portion of phase (1), called a nucleus, fluctuates to look like phase (2). The nucleus is at the barrier top in fig. 8.1. From this nucleus, phase (2) grows downhill in chemical potential if it is at lower free energy (P2). Thus the transition itself relies on microscopic fluctuations, and microscopic information is required to determine the barrier height, which is rather small. We need statistical mechanics to compute rates. |
If we are interested only in equilibrium, not how we get there, we can treat the phase transition like any other chemical reaction: \(A^{(1)}\) and \(A^{(2)}\) interconvert to yield mole numbers \(n^{(1)}_{eq}\) & \(n^{(2)}_{eq}\) or concentrations or pressures that minimize \(G\):
\[ A{(1)} \rightarrow A^{(2)} \]
\[G(T,P,n^{(i)})=\mu^{(1)}n^{(1)} +\mu^{(2)}n^{(2)} = \mu^{(1)}n^{(1)} + \mu^{(2)}(n-n^{(1)}) \]
where \(n\) is a constant. At equilibrium
\[ dG = 0\]
\[ \mu^{(1)} n^{(1)} +\mu^{(2)} n^{(2)} = (\mu^{(1)}-\mu^{(2)}) dn^{(2)} \]
\[ \mu^{(1)} = \mu^{(2)}\]
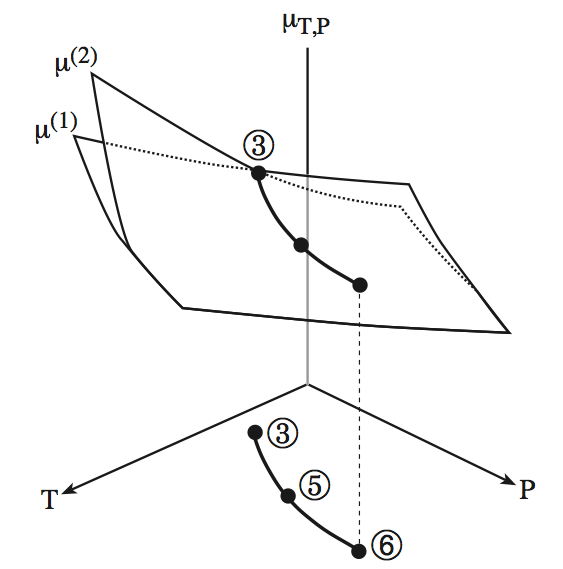
Fig. 8.2: Chemical potential of two phases as a function of temperature and pressure (Gibbs ensemble). At high \(T\), phase 1 is more stable, at (3)-(5) both phases coexist, at low \(T\) phase 2 is more stable. In this diagram at high T and P, the two chemical potentials become degenerate and only one phase exists at (6).
| Definition: First Order Phase Transition |
|---|
| A 1st order phase transition occurs when the chemical potential difference DG between two phases separated by a barrier vanishes. |
| Definition: Critical Phase Transition |
|---|
|
A critical phase transition occurs when the chemical potential barrier between two phases just vanishes. |