6.10: Molecular Orbitals
- Page ID
- 162848

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Before moving on to discuss methods that go beyond the HF model, it is appropriate to examine some of the computational effort that goes into carrying out a HF SCF calculation on a molecule. The primary differences that appear when molecules rather than atoms are considered are
- The electronic Hamiltonian \(H_e\) contains not only one nuclear-attraction Coulomb potential \(\sum_j Ze^2/r_j\), but a sum of such terms, one for each nucleus in the molecule:
\[\sum_a \sum_j \dfrac{Z_a e^2}{|r_j-R_a|}, \label{6.1.41}\]
whose locations are denoted \(R_a\).
- One has AO basis functions of the type discussed above located on each nucleus of the molecule. These functions are still denoted \(\chi_\mu(r-R_a)\), but their radial and angular dependences involve the distance and orientation of the electron relative to the particular nucleus on which the AO is located.
Other than these two changes, performing a SCF calculation on a molecule (or molecular ion) proceeds just as in the atomic case detailed earlier. Let us briefly review how this iterative process occurs.
Once atomic basis sets have been chosen for each atom, the one- and two-electron integrals appearing in the \(H_e\) and overlap matrices must be evaluated. There are numerous highly efficient computer codes that allow such integrals to be computed for \(s\), \(p\), \(d\), \(f\), and even \(g\), \(h\), and \(i\) basis functions. After executing one of these so-called integral packages for a basis with a total of \(M\) functions, one has available (usually on the computer's hard disk) of the order of \(\dfrac{M^2}{2}\) one-electron (\(\langle \chi_\mu | H_e | \chi_\nu \rangle\) and \(\langle \chi_\mu | \chi_\nu \rangle\)) and \(\dfrac{M^4}{8}\) two-electron (\(\langle \chi_\mu \chi_\delta | \chi_\nu \chi_\kappa \rangle\)) integrals. When treating extremely large atomic orbital basis sets (e.g., 500 or more basis functions), modern computer programs calculate the requisite integrals, but never store them on the disk. Instead, their contributions to the \(\langle\chi_\mu |H_e|\chi_\nu\rangle\) matrix elements are accumulated on the fly after which the integrals are discarded. This is usually referred to as the direct integral-driven approach.
Shapes, Sizes, and Energies of Orbitals
Each molecular spin-orbital (MO) that results from solving the HF SCF equations for a molecule or molecular ion consists of a sum of components involving all of the basis AOs:
\[\phi_j = \sum_\mu C_{J,\mu} \chi_\mu.\label{6.1.42}\]
In this expression, the \(C_{j,\mu}\) are referred to as LCAO-MO coefficients because they tell us how to linearly combine AOs to form the MOs. Because the AOs have various angular shapes (e.g., \(s\), \(p\), or \(d\) shapes) and radial extents (i.e., different orbital exponents), the MOs constructed from them can be of different shapes and radial sizes. Let’s look at a few examples to see what I mean.
The first example is rather simple and pertains to two H atoms combining to form the \(H_2\) molecule. The valence AOs on each H atom are the \(1s\) AOs; they combine to form the two valence MOs (\(\sigma\) and \(\sigma^*\)) depicted in Figure 6.1.4.
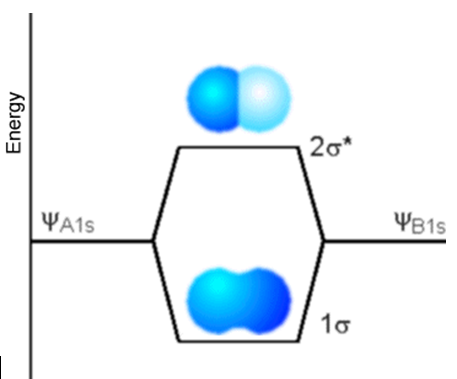
The bonding MO labeled s has LCAO-MO coefficients of equal sign for the two \(1s\) AOs, as a result of which this MO has the same sign near the left H nucleus (A) as near the right H nucleus (B). In contrast, the antibonding MO labeled \(\sigma^*\) has LCAO-MO coefficients of different sign for the A and B \(1s\) AOs. As was the case in the Hückel or tight-binding model outlined in Chapter 2, the energy splitting between the two MOs depends on the overlap \(\langle \chi_{1sA}|\chi_{1sB} \rangle\) between the two AOs which, in turn, depends on the distance \(R\) between the two nuclei.
An analogous pair of bonding and antibonding MOs arises when two \(p\) orbitals overlap sideways as in ethylene to form \(\pi\) and \(\pi^*\) MOs which are illustrated in Figure 6.1.5.
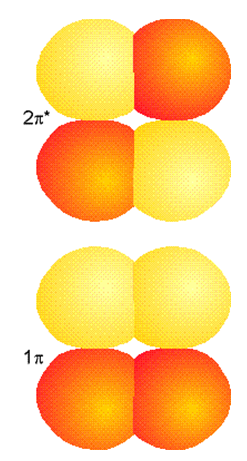
The shapes of these MOs clearly are dictated by the shapes of the AOs that comprise them and the relative signs of the LCAO-MO coefficients that relate the MOs to AOs. For the \(\pi\) MO, these coefficients have the same sign on the left and right atoms; for the \(\pi^*\) MO, they have opposite signs.
I should stress that the signs and magnitudes of the LCAO-MO coefficients arise as eigenvectors of the HF SCF matrix eigenvalue equation:
\[\sum_\mu \langle \chi_\nu|h_e| \chi_\mu \rangle C_{j,m} = \epsilon_j \sum_\mu \langle \chi_\nu|\chi_\mu \rangle C_{j,m} \]
It is a characteristic of such eigenvalue problems for the lower energy eigenfunctions to have fewer nodes than the higher energy solutions as we learned from several examples that we solved in Part 1 of this text.
Another thing to note about the MOs shown above is that they will differ in their quantitative details, but not in their overall shapes, when various functional groups are attached to the ethylene molecule’s C atoms. For example, if electron-withdrawing groups such as Cl, OH or Br are attached to one of the C atoms, the attractive potential experienced by a \(\pi\) electron near that C atom will be enhanced relative to the potential near the other C atom. As a result, the bonding MO will have larger LCAO-MO coefficients Ck,m belonging to tighter basis AOs \(\chi_\mu\) on this C atom. This will make the bonding \(\pi\) MO more radially compact in this region of space, although its nodal character and gross shape will not change. Alternatively, an electron donating group such as H3C- or t-butyl attached to one of the C centers will cause the \(\pi\) MO to be more diffuse (by making its LCAO-MO coefficients for more diffuse basis AOs larger).
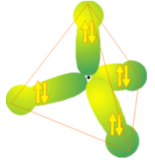
In addition to MOs formed primarily of AOs of one type (i.e., for \(H_2\) it is primarily s-type orbitals that form the \(\sigma\) and \(\sigma^*\) MOs; for ethylene’s \(\pi\) bond, it is primarily the C \(2p\) AOs that contribute), there are bonding and antibonding MOs formed by combining several AOs. For example, the four equivalent C-H bonding MOs in \(CH_4\) shown in Figure 6.1. 6 each involve C \(2s\) and \(2p\) as well as H \(1s\) basis AOs.
The energies of the MOs depend on two primary factors: the energies of the AOs from which the MOs are constructed and the overlap between these AOs. The pattern in energies for valence MOs formed by combining pairs of first-row atoms to form homo-nuclear diatomic molecules is shown in Figure 6.1. 7.
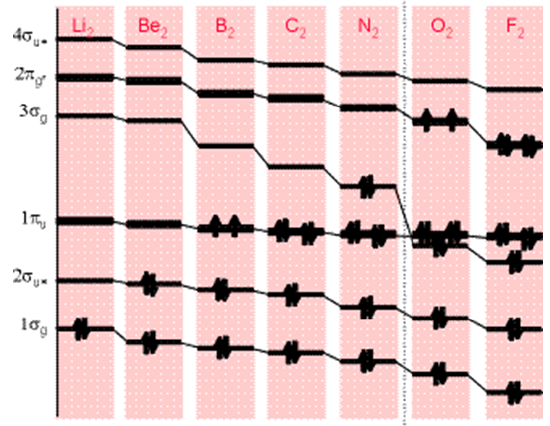
In this figure, the core MOs formed from the \(1s\) AOs are not shown; only those MOs formed from \(2s\) and \(2p\) AOs appear. The clear trend toward lower orbital energies as one moves from left to right is due primarily to the trends in orbital energies of the constituent AOs. That is, F being more electronegative than \(N\) has a lower-energy \(2p\) orbital than does \(N\).
Bonding, Anti-bonding, Non-bonding, and Rydberg Orbitals
As noted above, when valence AOs combine to form MOs, the relative signs of the combination coefficients determine, along with the AO overlap magnitudes, the MO’s energy and nodal properties. In addition to the bonding and antibonding MOs discussed and illustrated earlier, two other kinds of MOs are important to know about.
Non-bonding MOs arise, for example, when an orbital on one atom is not directed toward and overlapping with an orbital on a neighboring atom. For example, the lone pair orbitals on \(H_2O\) or on the oxygen atom of \(H_2C=O\) are non-bonding orbitals. They still are described in the LCAO-MO manner, but their \(\chi_{m,i}\) coefficients do not contain dominant contributions from more than one atomic center.
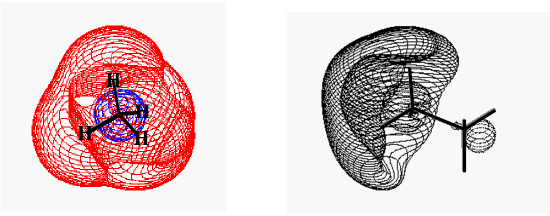
Finally, there is a type of orbital that all molecules possess but that is ignored in most elementary discussions of electronic structure. All molecules have so-called Rydberg orbitals. These orbitals can be thought of as large diffuse orbitals that describe the regions of space an electron would occupy if it were in the presence of the corresponding closed-shell molecular cation. Two examples of such Rydberg orbitals are shown in Figure 6.1.8. On the left, we see the Rydberg orbital of \(NH_4\) and on the right, that of \(H_3N-CH_3\). The former species can be thought of as a closed-shell ammonium cation \(NH_4^+\) around which a Rydberg orbital resides. The latter is protonated methyl amine with its Rydberg orbital.