
5.3: Chemical Change
- Page ID
- 11588

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Experimental Probes of Chemical Change
Many of the same tools that are used to determine the structures of molecules can also be used to follow the changes that the molecule undergoes as it is involved in a chemical reaction. Specifically, for any reaction in which one kind of molecule \(A\) is converted into another kind \(B\), one needs to have
- the ability to identify, via some physical measurement, the experimental signatures of both \(A\) and \(B\),
- the ability to relate the magnitude of these experimental signals to the concentrations \([A]\) and \([B]\) of these molecules, and
- the ability to monitor these signals as functions of time so that these concentrations can be followed as time evolves.
The third requirement is what allows one to determine the rates at which the \(A\) and \(B\) molecules are reacting.
Many of the experimental tools used to identify molecules (e.g., NMR allows one to identify functional groups and near-neighbor functional groups, IR also allows functional groups to be seen) and to determine their concentrations have restricted time scales over which they can be used. For example, NMR spectra require that the sample be studied for ca. 1 second or more to obtain a useable signal. Likewise, a mass spectroscopic analysis of a mixture of reacting species may require many second or minutes to carry out. These restrictions, in turn, limit the rates of reactions that can be followed using these experimental tools (e.g., one can not use NMR of mass spectroscopy to follow a reaction that occurs on a time scale of \(10^{-12}\) s).
Especially for very fast reactions and for reactions involving unstable species that can not easily be handled, so-called pump-probe experimental approaches are often used.
For example, suppose one were interested in studying the reaction of \(Cl\) radicals (e.g., as formed in the decomposition of chloroflurocarbons (CFCs) by ultraviolet light) with ozone to generate \(ClO\) and \(O_2\):
\[Cl + O_3 \rightarrow ClO + O_2^. \tag{5.3.1}\]
One can not simply deposit a known amount of \(Cl\) radicals from a vessel into a container in which gaseous \(O_3\) of a known concentration has been prepared; the \(Cl\) radicals will recombine and react with other species, making their concentrations difficult to determine. So, alternatively, one places known concentrations of some \(Cl\) radical precursor (e.g., a CFC or some other X-Cl species) and ozone into a reaction vessel. One then uses, for example, a very short light pulse whose photon's frequencies are tuned to a transition that will cause the X-Cl precursor to undergo rapid photodissociation:
\[h\nu + X-Cl \rightarrow X + Cl^.\tag{5.3.2}\]
Because the pump light source used to prepare the \(Cl\) radicals is of very short duration (\(\Delta{t}\)) and because the X-Cl dissociation is prompt, one knows, to within \(\Delta{t}\), the time at which the Cl radicals begin to react with the ozone. The initial concentration of the \(Cl\) radicals can be known if the quantum yield for the \(h\nu + X-Cl \rightarrow X + Cl\) reaction is known, This means that the intensity of photons, the probability of photon absorption by X-Cl, and the fraction of excited X-Cl molecules that dissociate to produce \(X + Cl\) must be known. Such information is available (albeit, from rather tedious earlier studies) for a variety of X-Cl precursors.
So, knowing the time at which the \(Cl\) radicals are formed and their initial concentrations, one then allows the \(Cl + O_3 h\nu \rightarrow ClO + O_2\) reaction to proceed for some time duration \(\Delta{t}\). One then, at \(t =\Delta{t}\), uses a second light source to probe either the concentration of the \(ClO\), the \(O_2\) or the \(O_3\), to determine the extent of progress of the reaction. Which species is so monitored depends on the availability of light sources whose frequencies these species absorb. Such probe experiments are carried out at a series of time delays \(\Delta{t}\), the result of which is the determination of the concentrations of some product or reactant species at various times after the initial pump event created the reactive \(Cl\) radicals. In this way, one can monitor, for example, the \(ClO\) concentration as a function of time after the \(Cl\) begins to react with the \(O_3\). If one has reason to believe that the reaction occurs in a single bimolecular event as
\[Cl + O_3 \rightarrow ClO + O_2 \tag{5.3.3}\]
one can then extract the rate constant k for the reaction by using the following kinetic scheme;
\[\dfrac{d[ClO]}{dt} = k [Cl] [O_3].\tag{5.3.4}\]
If the initial concentration of \(O_3\) is large compared to the amount of \(Cl\) that is formed in the pump event, \([O_3]\) can be taken as constant and known. If the initial concentration of \(Cl\) is denoted \([Cl]_0\), and the concentration of \(ClO\) is called \(x\), this kinetic equation reduces to
\[\dfrac{dx}{dt} = k ( [Cl]_0 -x) [O_3]\tag{5.3.5}\]
the solution of which is
\[[ClO] = x = [Cl]_0 (1 - \exp(-k[O_3]t)).\tag{5.3.6}\]
So, knowing the \([ClO]\) concentration as a function of time delay \(t\), and knowing the initial ozone concentration \([O_3]\) as well as the initial \(Cl\) radical concentration, one can find the rate constant \(k\).
Such pump-probe experiments are necessary when one wants to study species that must be generated and allowed to react immediately. This is essentially always the case when one or more of the reactants is a highly reactive species such as a radical. There is another kind of experiment that can be used to probe very fast reactions if the reaction and its reverse reaction can be brought into equilibrium to the extent that reactants and products both exist in measurable concentrations. For example, consider the reaction of an enzyme E and a substrate S to form the enzyme-substrate complex ES:
\[E + S \rightleftharpoons ES.\tag{5.3.7}\]
At equilibrium, the forward rate
\[k_f = [E]_{eq} [S]_{eq} \tag{5.3.8}\]
and the reverse rate
\[k_r = [ES]_{eq} \tag{5.3.9}\]
are equal:
\[k_f [E]_{eq} [S]_{eq} = k_r [ES]_{eq} \tag{5.3.10}\]
The idea behind so called perturbation techniques is to begin with a reaction that is in such an equilibrium condition and to then use some external means to slightly perturb the equilibrium. Because both the forward and reverse rates are assumed to be very fast, it is essential to use a perturbation that can alter the concentrations very quickly. This usually precludes simply adding a small amount of one or more of the reacting species to the reaction vessel. Instead, one might employ, for example, a fast light source or electric field pulse to perturb the equilibrium to one side or the other. For example, if the reaction thermochemistry is known, the equilibrium constant \(K_{eq}\) can be changed by rapidly heating the sample (e.g., with a fast laser pulse that is absorbed and rapidly heats the sample) and using
\[\dfrac{d \ln{K_{eq}}}{dT} = \dfrac{\Delta{H}}{RT^2} \tag{5.3.11}\]
to calculate the change in \(K_{eq}\) and thus the changes in concentrations caused by the sudden heating. Alternatively, if the polarity of the reactants and products is substantially different, one may use a rapidly applied electric field to quickly change the concentrations of the reactant and product species.
In such experiments, the concentrations of the species is shifted by a small amount \(\delta\) as a result of the application of the perturbation, so that
\[[ES] = [ES]_{eq} - \delta \tag{5.3.12}\]
\[[E] = [E]_{eq} + \delta \tag{5.3.13}\]
\[[S] = [S]_{eq} + \delta \tag{5.3.14}\]
once the perturbation has been applied and then turned off. Subsequently, the following rate law will govern the time evolution of the concentration change d:
\[- \dfrac{d\delta}{dt} = - k_r ([ES]_{eq} -\delta) + k_f ([E]_{eq} + \delta) ([S]_{eq} + \delta). \tag{5.3.15}\]
Assuming that \(\delta\) is very small (so that the term involving \(\delta^2\) cam be neglected) and using the fact that the forward and reverse rates balance at equilibrium, this equation for the time evolution of \(\delta\) can be reduced to:
\[- \dfrac{d\delta}{dt} = (k_r + k_f [S]_{eq} + k_f [E_{eq}]) d. v \tag{5.3.16}\]
So, the concentration deviations from equilibrium will return to equilibrium (i.e., \(\delta\) will decay to zero) exponentially with an effective rate coefficient that is equal to a sum of terms:
\[k_{eff} = k_r + k_f [S]_{eq} + k_f [E_{eq}] \tag{5.3.17}\]
involving both the forward and reverse rate constants.
So, by quickly perturbing an equilibrium reaction mixture for a short period of time and subsequently following the concentrations of the reactants or products as they return to their equilibrium values, one can extract the effective rate coefficient \(k_{eff}\). Doing this at a variety of different initial equilibrium concentrations (e,g., \([S]_{eq}\) and \([E]_{eq}\)), and seeing how \(k_{eff}\) changes, one can then determine both the forward and reverse rate constants.
Both the pump-probe and the perturbation methods require that one be able to quickly create (or perturb) concentrations of reactive species and that one have available an experimental probe that allows one to follow the concentrations of at least some of the species as time evolves. Clearly, for very fast reactions, this means that one must use experimental tools that can respond on a very short time scale. Modern laser technology and molecular beam methods have provided some of the most widely used of such tools. These experimental approaches are discussed in some detail in Chapter 8.
Theoretical Simulation of Chemical Change
The most common theoretical approach to simulating a chemical reaction is to use Newtonian dynamics to follow the motion of the nuclei on a Born-Oppenheimer electronic energy surface. If the molecule of interest contains few (\(N\)) atoms, such a surface could be computed (using the methods discussed in Chapter 6) at a large number of molecular geometries \(\{Q_K\}\) and then fit to an analytical function \(E(\{q_J\})\) of the \(3N-6\) or \(3N-5\) variables denoted \(\{q_J\}\). Knowing \(E\) as a function of these variables, one can then compute the forces
\[F_J = -\dfrac{\partial{E}}{\partial{q_J}} \tag{5.3.18}\]
along each coordinate, and then use the Newton equations
\[m_J \dfrac{d^2q_J}{dt^2} = F_J \tag{5.3.19}\]
to follow the time evolution of these coordinates and hence the progress of the reaction. The values of the coordinates \(\{q_J(t_L)\}\) at a series of discrete times \(t_L\) constitute what is called a classical trajectory. To simulate a chemical reaction, one begins the trajectory with initial coordinates characteristic of the reactant species (i.e., within one of the valleys on the reactant side of the potential surface) and one follows the trajectory long enough to determine whether the collision results in
- a non-reactive outcome characterized by final coordinates describing reactant not product molecules, or
- a reactive outcome that is recognized by the final coordinates describing product molecules rather than reactants.
One must do so for a large number of trajectories whose initial coordinates and moment are representative of the experimental conditions one is attempting to simulate. Then, one has to average the outcomes of these trajectories over this ensemble of initial conditions. More about how one carries out such ensemble averaging is discussed in Chapters 7 and 8.
If the molecule contains more than 3 or 4 atoms, it is more common to not compute the Born-Oppenheimer energy at a set of geometries and then fit this data to an analytical form. Instead, one begins a trajectory at some initial coordinates \(\{q_J(0)\}\) and with some initial momenta \(\{p_J(0)\}\) and then uses the Newton equations, usually in the finite-difference form:
\[q_J = q_J(0) + \dfrac{p_J(0)}{m_J} dt \tag{5.3.20}\]
\[p_J = p_J(0) -\dfrac{\partial E}{\partial q_J}(t=0) dt, \tag{5.3.21}\]
to propagate the coordinates and momenta forward in time by a small amount \(\delta{t}\). Here, \(\dfrac{\partial{E}}{\partial{q_J}}(t=0)\) denotes the gradient of the BO energy computed at the \(\{q_J(0)\}\) values of the coordinates. The above propagation procedure is then used again, but with the values of \(q_J\) and \(p_J\) appropriate to time \(t = \delta{t}\) as new initial coordinates and momenta, to generate yet another set of \(\{q_J\}\) and \(\{p_J\}\) values. In such direct dynamics approaches, the energy gradients, which produce the forces, are computed only at geometries that the classical trajectory encounters along its time propagation. In the earlier procedure, in which the BO energy is fit to an analytical form, one often computes \(E\) at geometries that the trajectory never accesses.
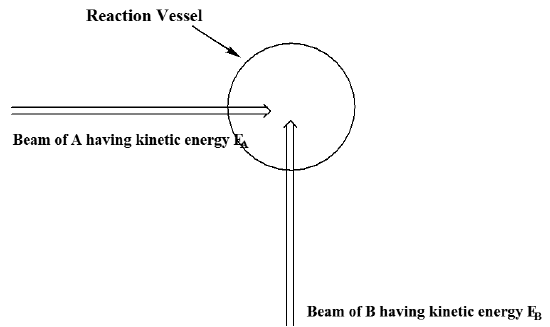
In carrying out such a classical trajectory simulation of a chemical reaction, there are other issues that must be addressed. In particular, as mentioned above, one can essentially never use any single trajectory to simulate a reaction carried out in a laboratory setting. One must perform a series of such trajectory calculations with a variety of different initial coordinates and momenta chosen in a manner to represent the experimental conditions of interest. For example, suppose one were to wish to model a molecular beam experiment in which a beam of species \(A\) having a well defined kinetic energy \(E_A\) collides with a beam of species \(B\) having kinetic energy \(E_B\) as shown in Figure 5.25.
Even though the \(A\) and \(B\) molecules all collide at right angles and with specified kinetic energies (and thus specified initial momenta), not all of these collisions occur head on. Figure 5.26 illustrates this point.
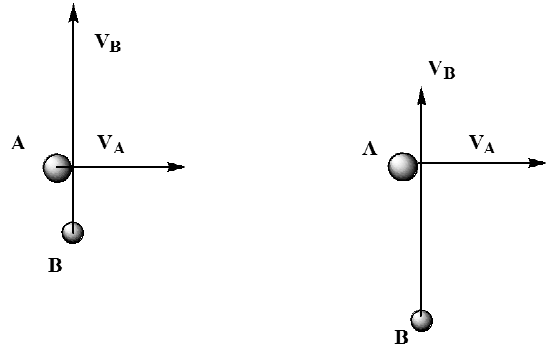
Here, we show two collisions between an \(A\) and a \(B\) molecule, both of which have identical \(A\) and \(B\) velocities \(V_A\) and \(V_B\), respectively. What differs in the two events is their distance of closest approach. In the collision shown on the left, the \(A\) and \(B\) come together closely. However, in the left collision, the A molecule is moving away from the region where \(B\) would strike it before \(B\) has reached it. These two cases can be viewed from a different perspective that helps to clarify their differences. In Figure 5.27, we illustrate these two collisions viewed from a frame of reference located on the \(A\) molecule.
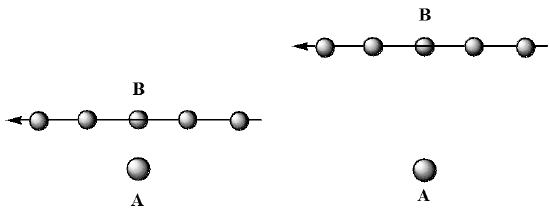
In this figure, we show the location of the \(B\) molecule relative to \(A\) at a series of times, showing \(B\) moving from right to left. In the figure on the left, the \(B\) molecule clearly undergoes a closer collision than is the case on the right. The distance of closest approach in each case is called the impact parameter and it represents the distance of closest approach if the colliding partners did not experience any attractive or repulsive interactions (as the above figures would be consistent with). Of course, when \(A\) and \(B\) have forces acting between them, the trajectories shown above would be modified to look more like those shown in Figure 5.28.
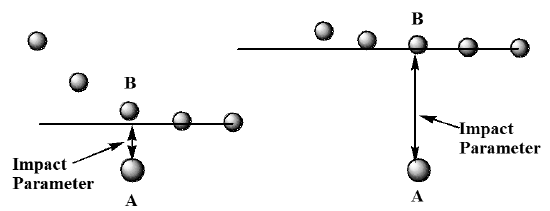
In both of these trajectories, repulsive intermolecular forces cause the trajectory to move away from its initial path, which defines the respective impact parameters.
So, even in this molecular beam example in which both colliding molecules have well specified velocities, one must carry out a number of classical trajectories, each with a different impact parameter b to simulate the laboratory event. In practice, the impact parameters can be chosen to range from \(b = 0\) (i.e., a head on collision) to some maximum value \(b_{Max}\) beyond which the \(A\) and \(B\) molecules no longer interact (and thus can no longer undergo reaction). Each trajectory is followed long enough to determine whether it leads to geometries characteristic of the product molecules. The fraction of such trajectories, weighted by the volume element \(2\pi b\,db\) for trajectories with impact parameters in the range between \(b\) and \(b + \delta{b}\), then gives the averaged fraction of trajectories that react.
In most simulations of chemical reactions, there are more initial conditions that also must sampled (i.e., trajectories with a variety of initial variables must be followed) and properly weighted. For example,
- if there is a range of velocities for the reactants \(A\) and/or \(B\), one must follow trajectories with velocities in this range and weigh the outcomes (i.e., reaction or not) of such trajectories appropriately (e.g., with a Maxwell-Boltzmann weighting factor), and
- if the reactant molecules have internal bond lengths, angles, and orientations, one must follow trajectories with different initial values of these variables and properly weigh each such trajectory (e.g., using the vibrational state's coordinate probability distribution as a weighting factor for the initial values of that coordinate).
As a result, to properly simulate a laboratory experiment of a chemical reaction, it usually requires one to follow a very large number of classical trajectories. Fortunately, such a task is well suited to distributed parallel computing, so it is currently feasible to do so even for rather complex reactions.
There is a situation in which the above classical trajectory approach can be foolish to pursue, even if there is reason to believe that a classical Newton description of the nuclear motions is adequate. This occurs when one has a rather high barrier to surmount to evolve from reactants to products and when the fraction of trajectories whose initial conditions permit this barrier to be accessed is very small. In such cases, one is faced with the reactive trajectories being very rare among the full ensemble of trajectories needed to properly simulate the laboratory experiment. Certainly, one can apply the trajectory-following technique outlined above, but if one observes, for example, that only one trajectory in 106 produces a reaction, one may not have adequate statistics to determine the reaction probability. One could subsequently run 108 trajectories (chosen again to represent the same experiment), and see whether 100 or 53 or 212 of these trajectories react, thereby increasing the precision of your reaction probability. However, it may be computationally impractical to perform 100 times as many trajectories to achieve better accuracy in the reaction probability.
When faced with such rare-event situations, one is usually better off using an approach that breaks the problem of determining what fraction of the (properly weighted) initial conditions produce reaction into two parts:
- among all of the (properly weighted) initial conditions, what fraction can access the high-energy barrier? and
- of those that do access the high barrier, how may react?
This way of formulating the reaction probability question leads to the transition state theory (TST) method that is treated in detail in Chapter 8, along with some of its more common variants.
Briefly, the answer to the first question posed above involves computing the quasi-equilibrium fraction of reacting species that reach the barrier region in terms of the partition functions of statistical mechanics. This step becomes practical if the chemical reactants can be assumed to be in some form of thermal equilibrium (which is where these kinds of models are useful). In the simplest form of TST, the answer to the second question posed above is taken to be "all trajectories that reach the barrier react". In more sophisticated variants, other models are introduced to take into consideration that not all trajectories that cross over the barrier indeed proceed onward to products and that some trajectories may tunnel through the barrier near its top. I will leave further discussion of the TST to Chapter 8.
In addition to the classical trajectory and TST approaches to simulating chemical reactions, there are more quantum approaches. These techniques should be used when the nuclei involved in the reaction include hydrogen or deuterium nuclei. A discussion of the details involved in quantum propagation is beyond the level of this Chapter, so I will delay it until Chapter 8.
Contributors and Attributions
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry
Integrated by Tomoyuki Hayashi (UC Davis)