
2.1: Free Electron Model of Polyenes
- Page ID
- 11571

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The particle-in-a-box type problems provide important models for several relevant chemical situations
The particle-in-a-box model for motion in one or two dimensions discussed earlier can obviously be extended to three dimensions. For two and three dimensions, it provides a crude but useful picture for electronic states on surfaces (i.e., when the electron can move freely on the surface but cannot escape to the vacuum or penetrate deeply into the solid) or in metallic crystals, respectively. I say metallic crystals because it is in such systems that the outermost valence electrons are reasonably well treated as moving freely rather than being tightly bound to a valence orbital on one of the constituent atoms or within chemical bonds localized to neighboring atoms.
Free motion within a spherical volume such as we discussed in Chapter 1 gives rise to eigenfunctions that are also used in nuclear physics to describe the motions of neutrons and protons in nuclei. In the so-called shell model of nuclei, the neutrons and protons fill separate \(s\), \(p\), \(d\), etc. orbitals (refer back to Chapter 1 to recall how these orbitals are expressed in terms of spherical Bessel functions and what their energies are) with each type of nucleon forced to obey the Pauli exclusion principle (i.e., to have no more than two nucleons in each orbital because protons and neutrons are Fermions). For example, \(^4He\) has two protons in \(1s\) orbitals and 2 neutrons in \(1s\) orbitals, whereas \(^3He\) has two \(1s\) protons and one \(1s\) neutron. To remind you, I display in Figure 2. 1 the angular shapes that characterize \(s\), \(p\), and \(d\) orbitals.
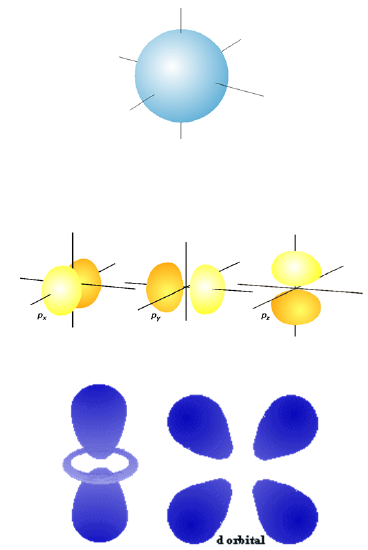
Figure 2.1. The angular shapes of \(s\), \(p\), and \(d\) functions
This same spherical box model has also been used to describe the valence electrons in quasi-spherical nano-clusters of metal atoms such as \(Cs_n\), \(Cu_n\), \(Na_n\), \(Au_n\), \(Ag_n\), and their positive and negative ions. Because of the metallic nature of these species, their valence electrons are essentially free to roam over the entire spherical volume of the cluster, which renders this simple model rather effective. In this model, one thinks of each valence electron being free to roam within a sphere of radius \(R\) (i.e., having a potential that is uniform within the sphere and infinite outside the sphere).
The orbitals that solve the Schrödinger equation inside such a spherical box are not the same in their radial shapes as the \(s\), \(p\), \(d\), etc. orbitals of atoms because, in atoms, there is an additional attractive Coulomb radial potential \(V(r) = -Ze^2/r\) present. In Chapter 1, we showed how the particle-in-a-sphere radial functions can be expressed in terms of spherical Bessel functions. In addition, the pattern of energy levels, which was shown in Chapter 1 to be related to the values of x at which the spherical Bessel functions \(j_L(x)\) vanish, are not the same as in atoms, again because the radial potentials differ. However, the angular shapes of the spherical box problem are the same as in atomic structure because, in both cases, the potential is independent of \(\theta\) and \(\phi\). As the orbital plots shown above indicate, the angular shapes of s, p, and \(d\) orbitals display varying number of nodal surfaces. The \(s\) orbitals have none, \(p\) orbitals have one, and \(d\) orbitals have two. Analogous to how the number of nodes related to the total energy of the particle constrained to the \(xy\) plane, the number of nodes in the angular wave functions indicates the amount of angular or orbital rotational energy. Orbitals of \(s\) shape have no angular energy, those of \(p\) shape have less then do \(d\) orbitals, etc.
It turns out that the pattern of energy levels derived from this particle-in-a-spherical-box model can offer reasonably accurate descriptions of what is observed experimentally. In particular, when a cluster (or cluster ion) has a closed-shell electronic configuration in which, for a given radial quantum number \(n\), all of the \(s\), \(p\), \(d\) orbitals associated with that \(n\) are doubly occupied, nanoscopic metal clusters are observed to display special stability (e.g., lack of chemical reactivity, large electron detachment energy). Clusters that produce such closed-shell electronic configurations are sometimes said to have magic-number sizes. The energy level expression given in Chapter 1
\[E_{L,n} = V_0 + (z_{L,n})^2 \dfrac{h^2}{2mR^2} \tag{2.1}\]
for an electron moving inside a sphere of radius \(R\) (and having a potential relative to the vacuum of \(V_0\)) can be used to model the energies of electron within metallic nano-clusters. Each electron occupies an orbital having quantum numbers \(n\), \(L\), and \(M\), with the energies of the orbitals given above in terms of the zeros \(\{z_{L,n}\}\) of the spherical Bessel functions. Spectral features of the nano-clusters are then determined by the energy gap between the highest occupied and lowest unoccupied orbital and can be tuned by changing the radius (\(R\)) of the cluster or the charge (i.e., number of electrons) of the cluster.
Another very useful application of the model problems treated in Chapter 1 is the one-dimensional particle-in-a-box, which provides a qualitatively correct picture for \(\pi\)-electron motion along the \(p_{\pi}\) orbitals of delocalized polyenes. The one Cartesian dimension corresponds to motion along the delocalized chain. In such a model, the box length \(L\) is related to the carbon-carbon bond length \(R\) and the number \(N\) of carbon centers involved in the delocalized network \(L=(N-1) R\). In Figure 2.2, such a conjugated network involving nine centers is depicted. In this example, the box length would be eight times the C-C bond length.
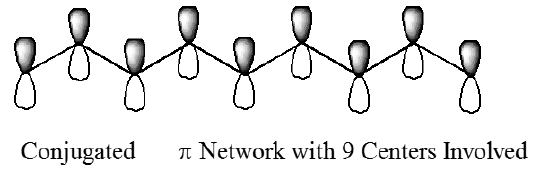
The eigenstates \(\psi_n(x)\) and their energies \(E_n\) represent orbitals into which electrons are placed. In the example case, if nine \(\pi\) electrons are present (e.g., as in the 1,3,5,7-nonatetraene radical), the ground electronic state would be represented by a total wave function consisting of a product in which the lowest four \(\psi\)'s are doubly occupied and the fifth \(\psi\) is singly occupied:
\[\Psi = \psi_1 \alpha\psi_1\beta \psi_2 \alpha \psi_2 \beta \psi_3 \alpha \psi_3\beta \psi_4 \alpha \psi_4 \beta \psi_5 \alpha. \tag{2.2}\]
The \(z\)-component spin angular momentum states of the electrons are labeled \(\alpha\) and \(\beta\) as discussed earlier.
We write the total wave function above as a product wave function because the total Hamiltonian involves the kinetic plus potential energies of nine electrons. To the extent that this total energy can be represented as the sum of nine separate energies, one for each electron, the Hamiltonian allows a separation of variables
\[H \cong \sum_{j=1}^9 H(j) \tag{2.3}\]
in which each H(j) describes the kinetic and potential energy of an individual electron. Of course, the full Hamiltonian contains electron-electron Coulomb interaction potentials \(e^2/r_{i,j}\) that cannot be written in this additive form. However, as we will treat in detail in Chapter 6, it is often possible to approximate these electron-electron interactions in a form that is additive.
Recall that when a partial differential equation has no operators that couple its different independent variables (i.e., when it is separable), one can use separation of variables methods to decompose its solutions into products. Thus, the (approximate) additivity of \(H\) implies that solutions of \(H \psi = E \psi\) are products of solutions to
\[H (j) \psi (\textbf{r}_j) = E_j \psi(\textbf{r}_j). \tag{2.4}\]
The two lowest \(\pi\pi^*\) excited states would correspond to states of the form
\[\psi^* = \psi_1\alpha \psi_1\beta \psi_2\alpha \psi_2\beta \psi_3\alpha \psi_3\beta \psi_4\alpha \psi_5\beta \psi_5\alpha, \tag{2.5a}\]
and
\[\psi'^* = \psi_1\alpha \psi_1\beta \psi_2\alpha \psi_2\beta \psi_3\alpha \psi_3\beta \psi_4\alpha \psi_4\beta \psi_6\alpha,\tag{2.5b}\]
where the spin-orbitals (orbitals multiplied by \(\alpha\) or \(\beta\)) appearing in the above products depend on the coordinates of the various electrons. For example,
\[\psi_1\alpha \psi_1\beta \psi_2\alpha \psi_2\beta \psi_3\alpha \psi_3\beta \psi_4\alpha \psi_5\beta \psi_5\alpha \tag{2.6a}\]
denotes
\[ \psi_1\alpha(\textbf{r}_1) \psi_1\beta (\textbf{r}_2) \psi_2\alpha (\textbf{r}_3) \psi_2\beta (\textbf{r}_4) \psi_3\alpha (\textbf{r}_5) \psi_3\beta (\textbf{r}_6) \psi_4a (\textbf{r}_7)\psi_5\beta (\textbf{r}_8) \psi_5\alpha (\textbf{r}_9). \tag{2.6b}\]
The electronic excitation energies from the ground state to each of the above excited states within this model would be
\[\Delta{E^*} = \dfrac{ \pi^2 \hbar^2}{2m} \left[ \dfrac{5^2}{L^2} - \dfrac{4^2}{L^2}\right] \tag{2.7a}\]
and
\[\Delta{E'^*} = \dfrac{ \pi^2 \hbar^2}{2m} \left[ \dfrac{6^2}{L^2} - \dfrac{5^2}{L^2}\right]. \tag{2.7b}\]
It turns out that this simple model of \(\pi\)-electron energies provides a qualitatively correct picture of such excitation energies. Its simplicity allows one, for example, to easily suggest how a molecule’s color (as reflected in the complementary color of the light the molecule absorbs) varies as the conjugation length \(L\) of the molecule varies. That is, longer conjugated molecules have lower-energy orbitals because \(L^2\) appears in the denominator of the energy expression. As a result, longer conjugated molecules absorb light of lower energy than do shorter molecules.
This simple particle-in-a-box model does not yield orbital energies that relate to ionization energies unless the potential inside the box is specified. Choosing the value of this potential \(V_0\) that exists within the box such that \(V_0 + \dfrac{\pi^2 \hbar^2}{2m} \dfrac{5^2}{L^2}\) is equal to minus the lowest ionization energy of the 1,3,5,7-nonatetraene radical, gives energy levels (as \(E = V_0 + \dfrac{\pi^2 \hbar^2}{2m} \dfrac{n^2}{L^2}\)), which can then be used as approximations to ionization energies.
The individual \(\pi\)-molecular orbitals
\[\psi_n = \sqrt{\dfrac{2}{L}} \sin\Big(\dfrac{n\pi x}{L}\Big) \tag{2.8}\]
are depicted in Figure 2.3 for a model of the 1,3,5 hexatriene \(\pi\)-orbital system for which the box length \(L\) is five times the distance \(R_{CC}\) between neighboring pairs of carbon atoms. The magnitude of the \(k^{th}\) C-atom centered atomic orbital in the \(n^{th}\) \(\pi\)-molecular orbital is given by
\[\sqrt{\dfrac{2}{L}} \sin\Big(\dfrac{n\pi(k-1)R_{CC}}{L}\Big).\]
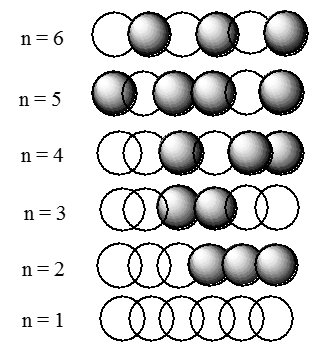
Figure 2.3. The phases of the six molecular orbitals of a chain containing six atoms.
In this figure, positive amplitude is denoted by the clear spheres, and negative amplitude is shown by the darkened spheres. Where two spheres of like shading overlap, the wave function has enhanced amplitude (i.e. there is a bonding interaction); where two spheres of different shading overlap, a node occurs (i.e., there is antibonding interaction). Once again, we note that the number of nodes increases as one ranges from the lowest-energy orbital to higher energy orbitals. The reader is once again encouraged to keep in mind this ubiquitous characteristic of quantum mechanical wave functions.
This simple model allows one to estimate spin densities at each carbon center and provides insight into which centers should be most amenable to electrophilic or nucleophilic attack. For example, radical attack at the \(C_5\) carbon of the nine-atom nonatetraene system described earlier would be more facile for the ground state \(\psi\) than for either \(\psi^*\) or \(\psi'^*\). In the former, the unpaired spin density resides in \(\psi_5\) (which varies as \(\sin(5\pi x/8R_{CC}\)) so is non-zero at \(x = L/2\)), which has non-zero amplitude at the \(C_5\) site \(x= L/2 = 4R_{CC}\). In \(\psi^*\) and \(\psi'*\), the unpaired density is in \(\psi_4\) and \(\psi_6\), respectively, both of which have zero density at \(C_5\) (because sin(npx/8RCC) vanishes for \(n = 4\) or \(6\) at \(x = 4R_{CC}\)). Plots of the wave functions for \(n\) ranging from 1 to 7 are shown in another format in Figure 2.4 where the nodal pattern is emphasized.
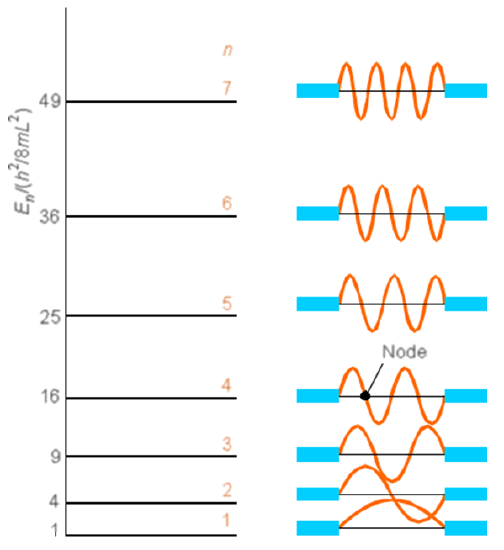
I hope that by now the student is not tempted to ask how the electron gets from one region of high amplitude, through a node, to another high-amplitude region. Remember, such questions are cast in classical Newtonian language and are not appropriate when addressing the wave-like properties of quantum mechanics.
Contributors and Attributions
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry
Integrated by Tomoyuki Hayashi (UC Davis)