
1.1: Why Quantum Mechanics is Necessary
- Page ID
- 11541

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The field of theoretical chemistry deals with the structures, bonding, reactivity, and physical properties of atoms, molecules, radicals, and ions all of whose sizes range from ca. 1 Å for atoms and small molecules to a few hundred Å for polymers and biological molecules such as DNA and proteins. Sometimes these building blocks combine to form nanoscopic materials (e.g., quantum dots, graphene sheets) whose dimensions span up to thousands of Å, making them amenable to detection using specialized microscopic tools. However, description of the motions and properties of the particles comprising such small systems has been found to not be amenable to treatment using classical mechanics. Their structures, energies, and other properties have only been successfully described within the framework of quantum mechanics. This is why quantum mechanics has to be mastered as part of learning theoretical chemistry.
We know that all molecules are made of atoms that, in turn, contain nuclei and electrons. As I discuss in this Chapter, the equations that govern the motions of electrons and of nuclei are not the familiar Newton equations
\[\textbf{F} = m \textbf{a} \tag{1.1}\]
but a new set of equations called Schrödinger equations. When scientists first studied the behavior of electrons and nuclei, they tried to interpret their experimental findings in terms of classical Newtonian motions, but such attempts eventually failed. They found that such small light particles behaved in a way that simply is not consistent with the Newton equations. Let me now illustrate some of the experimental data that gave rise to these paradoxes and show you how the scientists of those early times then used these data to suggest new equations that these particles might obey. I want to stress that the Schrödinger equation was not derived but postulated by these scientists. In fact, to date, to the best of my knowledge, no one has been able to derive the Schrödinger equation.
From the pioneering work of Bragg on diffraction of x-rays from planes of atoms or ions in crystals, it was known that peaks in the intensity of diffracted x-rays having wavelength l would occur at scattering angles q determined by the famous Bragg equation:
\[n \lambda = 2 d \sin{\theta} \tag{1.2}\]
where d is the spacing between neighboring planes of atoms or ions. These quantities are illustrated in Figure 1.1 shown below. There are may such diffraction peaks, each labeled by a different value of the integer \(n\) (\(n = 1, 2, 3, \cdots\)). The Bragg formula can be derived by considering when two photons, one scattering from the second plane in the figure and the second scattering from the third plane, will undergo constructive interference. This condition is met when the extra path length covered by the second photon (i.e., the length from points \(A\) to \(B\) to \(C\)) is an integer multiple of the wavelength of the photons.
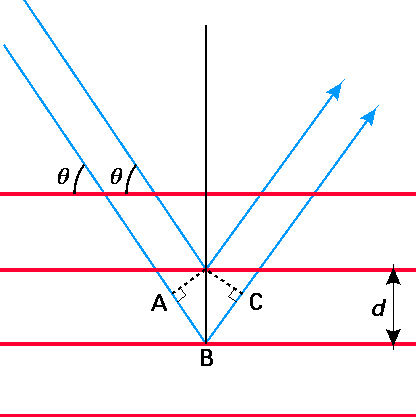
The importance of these x-ray scattering experiments to electrons and nuclei appears in the experiments of Davisson and Germer in 1927 who scattered electrons of (reasonably) fixed kinetic energy \(E\) from metallic crystals. These workers found that plots of the number of scattered electrons as a function of scattering angle \(\theta\) displayed peaks at angles \(\theta\) that obeyed a Bragg-like equation. The startling thing about this observation is that electrons are particles, yet the Bragg equation is based on the properties of waves. An important observation derived from the Davisson-Germer experiments was that the scattering angles \(\theta\) observed for electrons of kinetic energy \(E\) could be fit to the Bragg equation if a wavelength were ascribed to these electrons that was defined by
\[\lambda = \dfrac{h}{\sqrt{2m_e E}} \tag{1.3}\]
where \(m_e\) is the mass of the electron and h is the constant introduced by Max Planck and Albert Einstein in the early 1900s to relate a photon’s energy \(E\) to its frequency \(\nu\) via \(E = h\nu\)). These amazing findings were among the earliest to suggest that electrons, which had always been viewed as particles, might have some properties usually ascribed to waves. That is, as de Broglie has suggested in 1925, an electron seems to have a wavelength inversely related to its momentum, and to display wave-type diffraction. I should mention that analogous diffraction was also observed when other small light particles (e.g., protons, neutrons, nuclei, and small atomic ions) were scattered from crystal planes. In all such cases, Bragg-like diffraction is observed and the Bragg equation is found to govern the scattering angles if one assigns a wavelength to the scattering particle according to
\[\lambda = \dfrac{h}{\sqrt{2m E}} \tag{1.4}\]
where
- \(m\) is the mass of the scattered particle and
- \(h\) is Planck’s constant (6.62 x10-27 erg sec).
The observation that electrons and other small light particles display wave like behavior was important because these particles are what all atoms and molecules are made of. So, if we want to fully understand the motions and behavior of molecules, we must be sure that we can adequately describe such properties for their constituents. Because the classical Newtonian equations do not contain factors that suggest wave properties for electrons or nuclei moving freely in space, the above behaviors presented significant challenges.
Another problem that arose in early studies of atoms and molecules resulted from the study of the photons emitted from atoms and ions that had been heated or otherwise excited (e.g., by electric discharge). It was found that each kind of atom (i.e., H or C or O) emitted photons whose frequencies \(\nu\) were of very characteristic values. An example of such emission spectra is shown in Figure 1.2 for hydrogen atoms.
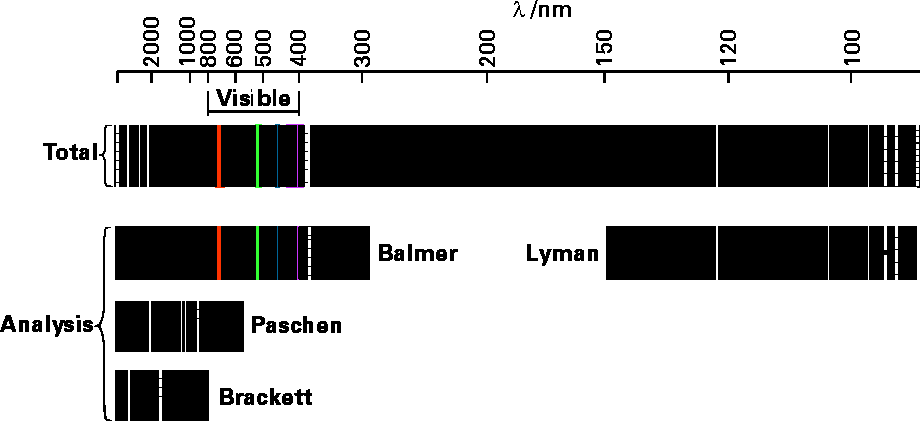
In the top panel, we see all of the lines emitted with their wave lengths indicated in nano-meters. The other panels show how these lines have been analyzed (by scientists whose names are associated) into patterns that relate to the specific energy levels between which transitions occur to emit the corresponding photons.
In the early attempts to rationalize such spectra in terms of electronic motions, one described an electron as moving about the atomic nuclei in circular orbits such as shown in Figure 1. 3.
A circular orbit was thought to be stable when the outward centrifugal force characterized by radius \(r\) and speed \(v\) (\(m_e v^2/r\)) on the electron perfectly counterbalanced the inward attractive Coulomb force (\(Ze^2/r^2\)) exerted by the nucleus of charge \(Z\):
\[m_e \dfrac{v^2}{r} = \dfrac{Ze^2}{r^2} \tag{1.5}\]
This equation, in turn, allows one to relate the kinetic energy \(\dfrac{1}{2} m_e v^2\) to the Coulombic energy \(Ze^2/r\), and thus to express the total energy \(E\) of an orbit in terms of the radius of the orbit:
\[E = \dfrac{1}{2} m_e v^2 – \dfrac{Ze^2}{r^2} = -\dfrac{1}{2} \dfrac{Ze^2}{r^2} \tag{1.6}\]
The energy characterizing an orbit or radius \(r\), relative to the \(E = 0\) reference of energy at \(r \rightarrow \infty\), becomes more and more negative (i.e., lower and lower) as \(r\) becomes smaller. This relationship between outward and inward forces allows one to conclude that the electron should move faster as it moves closer to the nucleus since \(v^2 = Ze^2/(r m_e)\). However, nowhere in this model is a concept that relates to the experimental fact that each atom emits only certain kinds of photons. It was believed that photon emission occurred when an electron moving in a larger circular orbit lost energy and moved to a smaller circular orbit. However, the Newtonian dynamics that produced the above equation would allow orbits of any radius, and hence any energy, to be followed. Thus, it would appear that the electron should be able to emit photons of any energy as it moved from orbit to orbit.
The breakthrough that allowed scientists such as Niels Bohr to apply the circular-orbit model to the observed spectral data involved first introducing the idea that the electron has a wavelength and that this wavelength l is related to its momentum by the de Broglie equation \(\lambda = h/p\). The key step in the Bohr model was to also specify that the radius of the circular orbit be such that the circumference of the circle \(2\pi r\) be equal to an integer (\(n\)) multiple of the wavelength \(\lambda\). Only in this way will the electron’s wave experience constructive interference as the electron orbits the nucleus. Thus, the Bohr relationship that is analogous to the Bragg equation that determines at what angles constructive interference can occur is
\[2 \pi r = n \lambda. \tag{1.7}\]
Both this equation and the analogous Bragg equation are illustrations of what we call boundary conditions; they are extra conditions placed on the wavelength to produce some desired character in the resultant wave (in these cases, constructive interference). Of course, there remains the question of why one must impose these extra conditions when the Newton dynamics do not require them. The resolution of this paradox is one of the things that quantum mechanics does.
Returning to the above analysis and using \(\lambda = h/p = h/(m_e v)\), \(2\pi r = n\lambda\), as well as the force-balance equation \(m_e v^2/r = Ze^2/r^2\), one can then solve for the radii that stable Bohr orbits obey:
\[r = \left(\dfrac{nh}{2\pi}\right)^2 \dfrac{1}{m_e Z e^2} \tag{1.8}\]
and, in turn for the velocities of electrons in these orbits
\[v = \dfrac{Z e^2}{nh/2\pi}. \tag{1.9}\]
These two results then allow one to express the sum of the kinetic (\(\dfrac{1}{2} m_e v^2\)) and Coulomb potential (\(-Ze^2/r\)) energies as
\[E = -\dfrac{1}{2} m_e Z^2 \dfrac{e^4}{(nh/2\pi)^2}. \tag{1.10}\]
Just as in the Bragg diffraction result, which specified at what angles special high intensities occurred in the scattering, there are many stable Bohr orbits, each labeled by a value of the integer \(n\). Those with small \(n\) have small radii (scaling as \(n^2\)), high velocities (scaling as 1/n) and more negative total energies (n.b., the reference zero of energy corresponds to the electron at \(r = \infty\), and with \(v = 0\)). So, it is the result that only certain orbits are allowed that causes only certain energies to occur and thus only certain energies to be observed in the emitted photons.
It turned out that the Bohr formula for the energy levels (labeled by \(n\)) of an electron moving about a nucleus could be used to explain the discrete line emission spectra of all one-electron atoms and ions (i.e., \(H\), \(He^+\), \(Li^{+2}\), etc., sometimes called hydrogenic species) to very high precision. In such an interpretation of the experimental data, one claims that a photon of energy
\[h\nu = R \left(\dfrac{1}{n_i^2} – \dfrac{1}{n_f^2}\right) \tag{1.11}\]
is emitted when the atom or ion undergoes a transition from an orbit having quantum number \(n_i\) to a lower-energy orbit having \(n_f\). Here the symbol \(R\) is used to denote the following collection of factors:
\[R = \dfrac{1}{2} m_e Z^2 \dfrac{e^4}{\Big(\dfrac{h}{2\pi}\Big)^2} \tag{1.12}\]
and is called the Rydberg unit of energy and is equal to 13.6 eV.
The Bohr formula for energy levels did not agree as well with the observed pattern of emission spectra for species containing more than a single electron. However, it does give a reasonable fit, for example, to the Na atom spectra if one examines only transitions involving only the single 3s valence electron. Moreover, it can be greatly improved if one introduces a modification designed to treat the penetration of the Na atom’s 3s and higher orbitals within the regions of space occupied by the 1s, 2s, and 2p orbitals. Such a modification to the Bohr model is achieved by introducing the idea of a so-called quantum defect d into the principal quantum number \(n\) so that the expression for the \(n\)-dependence of the orbitals changes to
\[E = \dfrac{-R}{(n-\delta)^2} \tag{1.13}\]
| Example 1.1 |
|---|
|
For example, choosing \(\delta\) equal to 0.41, 1.37, 2.23, 3.19, or 4.13 for Li, Na, K, Rb, and Cs, respectively, in this so-called Rydberg formula, one finds decent agreement between the \(n\)-dependence of the energy spacings of the singly excited valence states of these atoms. The fact that \(\delta\) is larger for Na than for Li and largest for Cs reflects that fact that the 3s orbital of Na penetrates the 1s, 2s, and 2p shells while the 2s orbital of Li penetrates only the 1s shell and the 6s orbital of Cs penetrates \(n = \) 1, 2, 3, 4, and 5 shells. |
It turns out this Rydberg formula can also be applied to certain electronic states of molecules. In particular, for closed-shell cations such as \(NH_4^+\), \(H_3O^+\), protonated alcohols and protonated amines (even on side chains of amino acids), an electron can be attached into a so-called Rydberg orbital to form corresponding neutral radicals such as \(NH_4\), \(H_3O\), \(R-NH_3\), or \(R-OH_2\). For example, in \(NH_4\), the electron bound to an underlying \(NH_4^+\) cation core. The lowest-energy state of this Rydberg species is often labeled 3s because \(NH_4^+\) is isoelectronic with the Na+ cation which binds an electron in its 3s orbital in its ground state. As in the cases of alkali atoms, these Rydberg molecules also possess excited electronic states. For example, the NH4 radical has states labeled 3p, 3d, 4s, 4p, 4d, 4f, etc. By making an appropriate choice of the quantum defect parameter d, the energy spacings among these states can be fit reasonably well to the Rydberg formula (Equation 1.13). In Figure 1.3.a several Rydberg orbitals of \(NH_4\) are shown
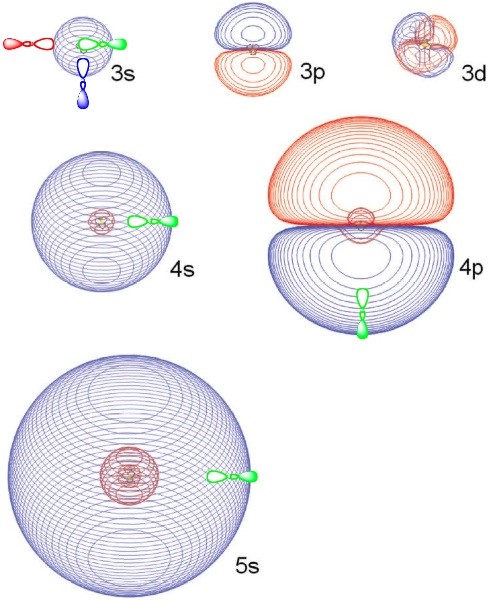
These Rydberg orbitals can be quite large (their sizes scale as \(n^2\), clearly have the s, p, or d angular shapes, and possess the expected number of radial nodes. However, for molecular Rydberg orbital’s, and unlike atomic Rydberg orbitals, the three \(p\), five \(d\), seven \(f\), etc. orbitals are not degenerate; instead they are split in energy in a manner reflecting the symmetry of the underlying cation’s symmetry. For example, for \(NH_4\), the three \(3p\) orbitals are degenerate and belong to \(t_2\) symmetry in the \(T_d\) point group; the five \(3d\) orbitals are split into three degenerate \(t_2\) and two degenerate e orbitals.
So, the Bohr model works well for one-electron atoms or ions and the quantum defect-modified Bohr equation describes reasonably well some states of alkali atoms and of Rydberg molecules. The primary reason for the breakdown of the Bohr formula is the neglect of electron-electron Coulomb repulsions in its derivation, which are qualitatively corrected for by using the quantum defect parameter for Rydberg atoms and molecules. Nevertheless, the success of the Bohr model made it clear that discrete emission spectra could only be explained by introducing the concept that not all orbits were allowed. Only special orbits that obeyed a constructive-interference condition were really accessible to the electron’s motions. This idea that not all energies were allowed, but only certain quantized energies could occur was essential to achieving even a qualitative sense of agreement with the experimental fact that emission spectra were discrete.
In summary, two experimental observations on the behavior of electrons that were crucial to the abandonment of Newtonian dynamics were the observations of electron diffraction and of discrete emission spectra. Both of these findings seem to suggest that electrons have some wave characteristics and that these waves have only certain allowed (i.e., quantized) wavelengths.
So, now we have some idea about why Newton’s equations fail to account for the dynamical motions of light and small particles such as electrons and nuclei. We see that extra conditions (e.g., the Bragg condition or constraints on the de Broglie wavelength) could be imposed to achieve some degree of agreement with experimental observation. However, we still are left wondering what equations can be applied to properly describe such motions and why the extra conditions are needed. It turns out that a new kind of equation based on combining wave and particle properties needed to be developed to address such issues. These are the so-called Schrödinger equations to which we now turn our attention.
As I said earlier, no one has yet shown that the Schrödinger equation follows deductively from some more fundamental theory. That is, scientists did not derive this equation; they postulated it. Some idea of how the scientists of that era dreamed up the Schrödinger equation can be had by examining the time and spatial dependence that characterizes so-called traveling waves. It should be noted that the people who worked on these problems knew a great deal about waves (e.g., sound waves and water waves) and the equations they obeyed. Moreover, they knew that waves could sometimes display the characteristic of quantized wavelengths or frequencies (e.g., fundamentals and overtones in sound waves). They knew, for example, that waves in one dimension that are constrained at two points (e.g., a violin string held fixed at two ends) undergo oscillatory motion in space and time with characteristic frequencies and wavelengths. For example, the motion of the violin string just mentioned can be described as having an amplitude \(A(x,t)\) at a position \(x\) along its length at time \(t\) given by
\[A(x,t) = A(x,o) \cos(2\pi \nu t), \tag{1.14}\]
where \(nu\) is its oscillation frequency. The amplitude’s spatial dependence also has a sinusoidal dependence given by
\[A(x,0) = A \sin (\dfrac{2\pi x}{\lambda}) \tag{1.15}\]
where \(\lambda\) is the crest-to-crest length of the wave. Two examples of such waves in one dimension are shown in Figure 1. 4.
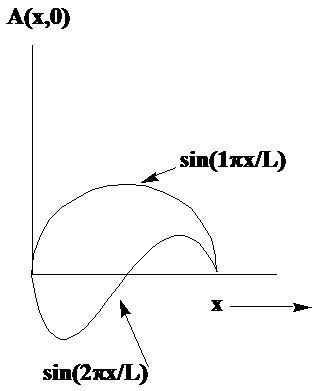
In these cases, the string is fixed at \(x = 0\) and at \(x = L\), so the wavelengths belonging to the two waves shown are \(\lambda = 2L\) and \(\lambda = L\). If the violin string were not clamped at \(x = L\), the waves could have any value of \(\lambda\). However, because the string is attached at \(x = L\), the allowed wavelengths are quantized to obey
\[\lambda = \dfrac{2L}{n}, \tag{1.16}\]
where \(n = 1, 2, 3, 4, \cdots\). The equation that such waves obey, called the wave equation, reads:
\[\dfrac{d^2A(x,t)}{dt^2} = c^2 \dfrac{d^2A}{dx^2} \tag{1.17}\]
where \(c\) is the speed at which the wave travels. This speed depends on the composition of the material from which the violin string is made; stiff string material produces waves with higher speeds than for softer material. Using the earlier expressions for the \(x-\) and \(t\)-dependences of the wave \(A(x,t)\), we find that the wave’s frequency and wavelength are related by the so-called dispersion equation:
\[\nu^2 = \left(\dfrac{c}{\lambda}\right)^2, \tag{1.18}\]
or
\[c = \lambda \nu. \tag{1.19}\]
This relationship implies, for example, that an instrument string made of a very stiff material (large \(c\)) will produce a higher frequency tone for a given wavelength (i.e., a given value of \(n\)) than will a string made of a softer material (smaller \(c\)).
For waves moving on the surface of, for example, a rectangular two-dimensional surface of lengths \(L_x\) and \(L_y\), one finds
\[A(x,y,t) = \sin \left(n_x \dfrac{p_x}{L_x}\right) \sin\left(n_y \dfrac{p_y}{L_y}\right) \cos\left(2\pi \nu t\right). \tag{1.20}\]
Hence, the waves are quantized in two dimensions because their wavelengths must be constrained to cause \(A(x,y,t)\) to vanish at \(x = 0\) and \(x = L_x\) as well as at \(y = 0\) and \(y = L_y\) for all times \(t\).
It is important to note, in closing this discussion of waves on strings and surfaces, that it is not being a solution to the Schrödinger equation that results in quantization of the wavelengths. Instead, it is the condition that the wave vanish at the boundaries that generates the quantization. You will see this trend time and again throughout this text; when a wave function is subject to specific constraints at its inner or outer boundary (or both), quantization will result; if these boundary conditions are not present, quantization will not occur. Let us now return to the issue of waves that describe electrons moving.
The pioneers of quantum mechanics examined functional forms similar to those shown above. For example, forms such as \(A = \exp[\pm 2\pi(\nu t – x/\lambda)]\) were considered because they correspond to periodic waves that evolve in \(x\) and \(t\) under no external \(x\)- or \(t\)-dependent forces. Noticing that
\[\dfrac{d^2A}{dx^2} = - \left(\dfrac{2\pi}{\lambda} \right)^2 A \tag{1.21}\]
and using the de Broglie hypothesis \(\lambda = h/p\) in the above equation, one finds
\[\dfrac{d^2A}{dx^2} = - p^2 \Big(\dfrac{2\pi}{h}\Big)^2 A. \tag{1.22}\]
If \(A\) is supposed to relate to the motion of a particle of momentum p under no external forces (since the waveform corresponds to this case), \(p^2\) can be related to the energy \(E\) of the particle by \(E = p^2/2m\). So, the equation for \(A\) can be rewritten as:
\[\dfrac{d^2A}{dx^2} = - 2m E \Big(\dfrac{2\pi}{h}\Big)^2 A, \tag{1.23}\]
or, alternatively,
\[- \Big(\dfrac{h}{2\pi}\Big)^2 (\dfrac{1}{2}m) \dfrac{d^2A}{dx^2} = E A. \tag{1.23}\]
Returning to the time-dependence of \(A(x,t)\) and using \(\nu = E/h\), one can also show that
\[i \Big(\dfrac{h}{2\pi}\Big) \dfrac{dA}{dt} = E A, \tag{1.24}\]
which, using the first result, suggests that
\[i \Big(\dfrac{h}{2\pi}\Big) \dfrac{dA}{dt} = - \Big(\dfrac{h}{2\pi}\Big)^2 (\dfrac{1}{2}m) \dfrac{d^2A}{dx^2}. \tag{1.25}\]
This is a primitive form of the Schrödinger equation that we will address in much more detail below. Briefly, what is important to keep in mind that the use of the de Broglie and Planck/Einstein connections (\(\lambda = h/p\) and \(E = h\nu\)), both of which involve the constant h, produces suggestive connections between
\[i \Big(\dfrac{h}{2\pi}\Big) \dfrac{d}{dt} \text{ and } E \tag{1.26}\]
and between
\[p^2 \text {and } – \Big(\dfrac{h}{2\pi}\Big)^2 \dfrac{d^2}{dx^2} \tag{1.27}\]
or, alternatively, between
\[p \text{ and } – i \Big(\dfrac{h}{2\pi}\Big) \dfrac{d}{dx}.\tag{1.28}\]
These connections between physical properties (energy \(E\) and momentum \(p\)) and differential operators are some of the unusual features of quantum mechanics.
The above discussion about waves and quantized wavelengths as well as the observations about the wave equation and differential operators are not meant to provide or even suggest a derivation of the Schrödinger equation. Again the scientists who invented quantum mechanics did not derive its working equations. Instead, the equations and rules of quantum mechanics have been postulated and designed to be consistent with laboratory observations. My students often find this to be disconcerting because they are hoping and searching for an underlying fundamental basis from which the basic laws of quantum mechanics follow logically. I try to remind them that this is not how theory works. Instead, one uses experimental observation to postulate a rule or equation or theory, and one then tests the theory by making predictions that can be tested by further experiments. If the theory fails, it must be refined, and this process continues until one has a better and better theory. In this sense, quantum mechanics, with all of its unusual mathematical constructs and rules, should be viewed as arising from the imaginations of scientists who tried to invent a theory that was consistent with experimental data and which could be used to predict things that could then be tested in the laboratory. Thus far, this theory has proven to be reliable, but, of course, we are always searching for a new and improved theory that describes how small light particles move.
If it helps you to be more accepting of quantum theory, I should point out that the quantum description of particles reduces to the classical Newton description under certain circumstances. In particular, when treating heavy particles (e.g., macroscopic masses and even heavier atoms), it is often possible to use Newton dynamics. Soon, we will discuss in more detail how the quantum and classical dynamics sometimes coincide (in which case one is free to use the simpler Newton dynamics). So, let us now move on to look at this strange Schrödinger equation that we have been digressing about for so long.
Contributors and Attributions
Jack Simons (Henry Eyring Scientist and Professor of Chemistry, U. Utah) Telluride Schools on Theoretical Chemistry
Integrated by Tomoyuki Hayashi (UC Davis)