
Reactions of Substituent Groups
- Page ID
- 1207

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Oxidation of Alkyl Side-Chains
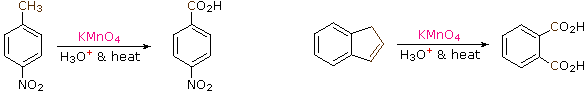
The benzylic hydrogens of alkyl substituents on a benzene ring are activated toward free radical attack, as noted earlier. Furthermore, SN1, SN2 and E1 reactions of benzylic halides, show enhanced reactivity, due to the adjacent aromatic ring. The possibility that these observations reflect a general benzylic activation is supported by the susceptibility of alkyl side-chains to oxidative degradation, as shown in the following examples (the oxidized side chain is colored). Such oxidations are normally effected by hot acidic pemanganate solutions, but for large scale industrial operations catalyzed air-oxidations are preferred. Interestingly, if the benzylic position is completely substituted this oxidative degradation does not occur (second equation, the substituted benzylic carbon is colored blue).
| C6H5–CH2CH2CH2CH3 + KMnO4 + H3O(+) & heat | C6H5–CO2H + CO2 | |
| p-(CH3)3C–C6H4–CH3 + KMnO4 + H3O(+) & heat | p-(CH3)3C–C6H4–CO2H |
These equations are not balanced. The permanganate oxidant is reduced, usually to Mn(IV) or Mn(II). Two other examples of this reaction are given below, and illustrate its usefulness in preparing substituted benzoic acids.
Reduction of Nitro Groups and Aryl Ketones
Electrophilic nitration and Friedel-Crafts acylation reactions introduce deactivating, meta-directing substituents on an aromatic ring. The attached atoms are in a high oxidation state, and their reduction converts these electron withdrawing functions into electron donating amino and alkyl groups. Reduction is easily achieved either by catalytic hydrogenation (H2 + catalyst), or with reducing metals in acid. Examples of these reductions are shown here, equation 6 demonstrating the simultaneous reduction of both functions. Note that the butylbenzene product in equation 4 cannot be generated by direct Friedel-Crafts alkylation due to carbocation rearrangement. The zinc used in ketone reductions, such as 5, is usually activated by alloying with mercury (a process known as amalgamation).
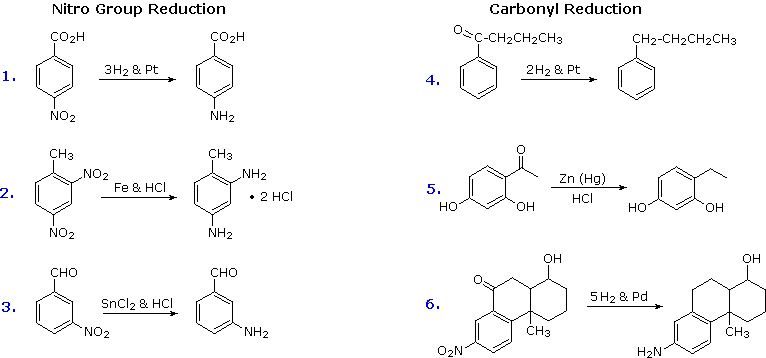
Several alternative methods for reducing nitro groups to amines are known. These include zinc or tin in dilute mineral acid, and sodium sulfide in ammonium hydroxide solution. The procedures described above are sufficient for most cases.
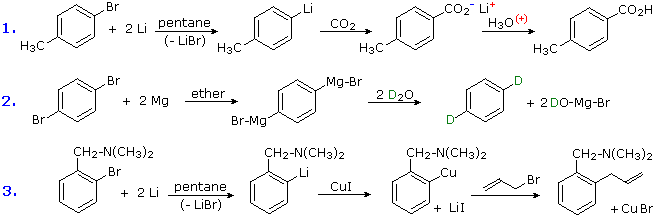
Conversion of Halogens to Organometallic Reagents
The reaction of alkyl and aryl halides with reactive metals (usually Li & Mg) to give nucleophilic reagents has been noted. This provides a powerful tool for the conversion of chloro, bromo or iodo substituents into a variety of other groups. Many reactions of these aryl lithium and Grignard reagents will be discussed in later sections, and the following equations provide typical examples of carboxylation, protonation and Gilman coupling. Metal halogen exchange reactions take place at low temperature, and may be used to introduce iodine at designated locations. An example of this method will be displayed below by clicking on the diagram. In this example care must be taken to maintain a low temperature, because elimination to an aryne intermediate takes place on warming.
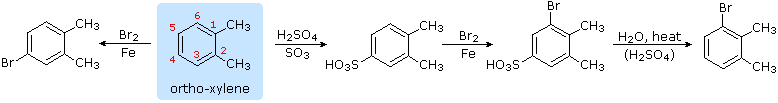
Hydrolysis of Sulfonic Acids
The potential reversibility of the aromatic sulfonation reaction was noted earlier. The following equation illustrates how this characteristic of the sulfonic acids may be used to prepare the 3-bromo derivative of ortho-xylene. Direct bromination would give the 4-bromo derivative.
Modifying the Influence of Strong Activating Groups
The strongest activating and ortho/para-directing substituents are the amino (-NH2) and hydroxyl (-OH) groups. Direct nitration of phenol (hydroxybenzene) by dilute nitric acid gives modest yields of nitrated phenols and considerable oxidative decomposition to tarry materials; aniline (aminobenzene) is largely destroyed. Bromination of both phenol and aniline is difficult to control, with di- and tri-bromo products forming readily. Because of their high nucleophilic reactivity, aniline and phenol undergo substitution reactions with iodine, a halogen that is normally unreactive with benzene derivatives. The mixed halogen iodine chloride (ICl) provides a more electrophilic iodine moiety, and is effective in iodinating aromatic rings having less powerful activating substituents.
| C6H5–NH2 + I2 + NaHCO3 | p-I–C6H4–NH2 + NaI + CO2 + H2O |
By acetylating the heteroatom substituent on phenol and aniline, its activating influence can be substantially attenuated. For example, acetylation of aniline gives acetanilide (first step in the following equation), which undergoes nitration at low temperature, yielding the para-nitro product in high yield. The modifying acetyl group can then be removed by acid-catalyzed hydrolysis (last step), to yield para-nitroaniline. Although the activating influence of the amino group has been reduced by this procedure, the acetyl derivative remains an ortho/para-directing and activating substituent.
C6H5–NH2 + (CH3CO)2O |
pyridine (a base) |
C6H5–NHCOCH3 |
HNO3 , 5 ºC |
p-O2N–C6H4–NHCOCH3 |
H3O(+) & heat |
p-O2N–C6H4–NH2 |
The following diagram illustrates how the acetyl group acts to attenuate the overall electron donating character of oxygen and nitrogen. The non-bonding valence electron pairs that are responsible for the high reactivity of these compounds (blue arrows) are diverted to the adjacent carbonyl group (green arrows). However, the overall influence of the modified substituent is still activating and ortho/para-directing.
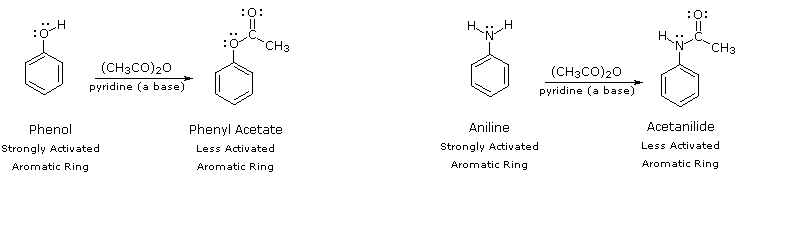 |
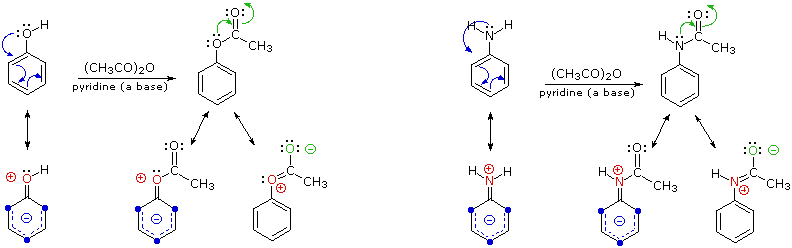
Deactivation of Phenol to Phenyl Acetate and Aniline to Acetanilide
Contributors
- William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry