
Characteristics of Specific Substitution Reactions of Benzenes
- Page ID
- 1202

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The conditions commonly used for the aromatic substitution reactions discussed here are repeated in the table below. The electrophilic reactivity of these different reagents varies. We find, for example, that nitration of nitrobenzene occurs smoothly at 95 ºC, giving meta-dinitrobenzene, whereas bromination of nitrobenzene (ferric catalyst) requires a temperature of 140 ºC. Also, as noted earlier, toluene undergoes nitration about 25 times faster than benzene, but chlorination of toluene is over 500 times faster than that of benzene. From this we may conclude that the nitration reagent is more reactive and less selective than the halogenation reagents.
Both sulfonation and nitration yield water as a by-product. This does not significantly affect the nitration reaction (note the presence of sulfuric acid as a dehydrating agent), but sulfonation is reversible and is driven to completion by addition of sulfur trioxide, which converts the water to sulfuric acid. The reversibility of the sulfonation reaction is occasionally useful for removing this functional group.
| Halogenation: | C6H6 | + Cl2 & heat FeCl3 catalyst |
——> | C6H5Cl Chlorobenzene |
+ HCl |
| Nitration: | C6H6 | + HNO3 & heat H2SO4 catalyst |
——> | C6H5NO2 Nitrobenzene |
+ H2O |
| Sulfonation: | C6H6 | + H2SO4 + SO3 & heat |
——> | C6H5SO3H Benzenesulfonic acid |
+ H2O |
| Alkylation: Friedel-Crafts |
C6H6 | + R-Cl & heat AlCl3 catalyst |
——> | C6H5-R An Arene |
+ HCl |
| Acylation: Friedel-Crafts |
C6H6 | + RCOCl & heat AlCl3 catalyst |
——> | C6H5COR An Aryl Ketone |
+ HCl |
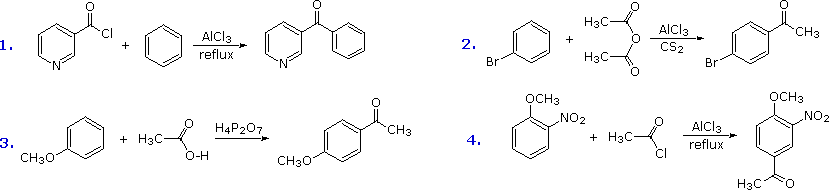
The Friedel-Crafts acylation reagent is normally composed of an acyl halide or anhydride mixed with a Lewis acid catalyst such as AlCl3. This produces an acylium cation, R-C≡O(+), or a related species. Such electrophiles are not exceptionally reactive, so the acylation reaction is generally restricted to aromatic systems that are at least as reactive as chlorobenzene. Carbon disulfide is often used as a solvent, since it is unreactive and is easily removed from the product. If the substrate is a very reactive benzene derivative, such as anisole, carboxylic esters or acids may be the source of the acylating electrophile. Some examples of Friedel-Crafts acylation reactions are shown in the following diagram. The first demonstrates that unusual acylating agents may be used as reactants. The second makes use of an anhydride acylating reagent, and the third illustrates the ease with which anisole reacts, as noted earlier. The H4P2O7 reagent used here is an anhydride of phosphoric acid called pyrophosphoric acid. Finally, the fourth example illustrates several important points. Since the nitro group is a powerful deactivating substituent, Friedel-Crafts acylation of nitrobenzene does not take place under any conditions. However, the presence of a second strongly-activating substituent group permits acylation; the site of reaction is that favored by both substituents.
A common characteristic of the halogenation, nitration, sulfonation and acylation reactions is that they introduce a deactivating substituent on the benzene ring. As a result, we do not normally have to worry about disubstitution products being formed. Friedel-Crafts alkylation, on the other hand, introduces an activating substituent (an alkyl group), so more than one substitution may take place. If benzene is to be alkylated, as in the following synthesis of tert-butylbenzene, the mono-alkylated product is favored by using a large excess of this reactant. When the molar ratio of benzene to alkyl halide falls below 1:1, para-ditert-butylbenzene becomes the major product.
C6H6 (large excess) + (CH3)3C-Cl + AlCl3 ——> C6H5-C(CH3)3 + HCl
The carbocation electrophiles required for alkylation may be generated from alkyl halides (as above), alkenes + strong acid or alcohols + strong acid. Since 1º-carbocations are prone to rearrangement, it is usually not possible to introduce 1º-alkyl substituents larger than ethyl by Friedel-Crafts alkylation. For example, reaction of excess benzene with 1-chloropropane and aluminum chloride gives a good yield of isopropylbenzene (cumene).
C6H6 (large excess) + CH3CH2CH2-Cl + AlCl3 ——> C6H5-CH(CH3)2 + HClAdditional examples of Friedel-Crafts alkylation reactions are shown in the following diagram.
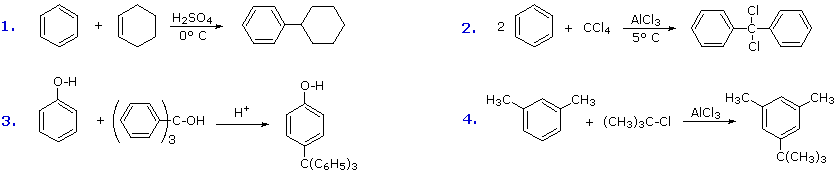
The first and third examples show how alkenes and alcohols may be the source of the electrophilic carbocation reactant. The triphenylmethyl cation generated in the third case is relatively unreactive, due to extensive resonance charge delocalization, and only substitutes highly activated aromatic rings. The second example shows an interesting case in which a polychlororeactant is used as the alkylating agent. A four fold excess of carbon tetrachloride is used to avoid tri-alkylation of this reagent, a process that is retarded by steric hindrance. The fourth example illustrates the poor orientational selectivity often found in alkylation reactions of activated benzene rings. The bulky tert-butyl group ends up attached to the reactive meta-xylene ring at the least hindered site. This may not be the site of initial bonding, since polyalkylbenzenes rearrange under Friedel-Crafts conditions (para-dipropylbenzene rearranges to meta-dipropylbenzene on heating with AlCl3).
A practical concern in the use of electrophilic aromatic substitution reactions in synthesis is the separation of isomer mixtures. This is particularly true for cases of ortho-para substitution, which often produce significant amounts of the minor isomer. As a rule, para-isomers predominate except for some reactions of toluene and related alkyl benzenes. Separation of these mixtures is aided by the fact that para-isomers have significantly higher melting points than their ortho counterparts; consequently, fractional crystallization is often an effective isolation technique. Since meta-substitution favors a single product, separation of trace isomers is normally not a problem.
Contributors
- William Reusch, Professor Emeritus (Michigan State U.), Virtual Textbook of Organic Chemistry