
II. Intermolecular Addition Reactions
- Page ID
- 23951
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)A. General Reaction Equation
A useful terminology for describing radical addition reactions is given in eq 1. According to this description, when a carbon-centered radical reacts with a carbon–carbon double bond, it adds to the β-carbon atom and creates a new radical center on the α-carbon atom. The letters X, Y, and Z in eq 1 represent substituents attached to the three carbon atoms directly involved in the reaction.
.png?revision=1&size=bestfit&width=375&height=127)
B. Reaction at the Less-Substituted Carbon Atom
A characteristic of radical addition reactions is that a carbon-centered radical adds regioselectively to the less-substituted atom in a C–C multiple bond.7–10 The reaction shown in Scheme 1 provides a typical example. Other reactions involving double bonds with different substituents (eq 2)11 and double bonds with more than one substituent (eq 3)12.13 exhibit similar regioselectivity. Explaining regioselectivity in addition reactions begins by noting that they usually are not reversible;14 therefore, information about transition-state structures is critical to understanding the selectivity in these kinetically controlled reactions.
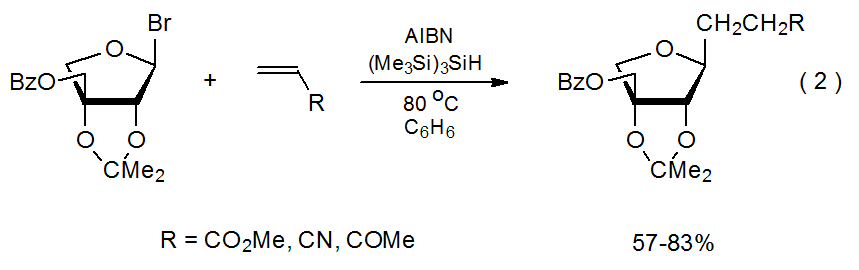.png?revision=1&size=bestfit&width=430&height=135)
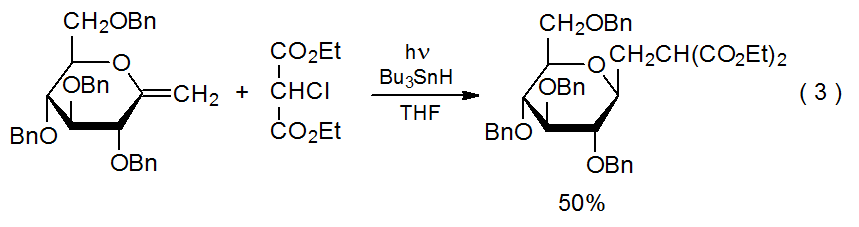.png?revision=1&size=bestfit&width=430&height=114)
C. Transition-State Structure
The structure for the transition state in a radical addition reaction, as determined from molecular-orbital calculations, is shown in Figure 1.8 Several aspects of this structure affect reaction regioselectivity. The first is that the structure is unsymmetrical.7,8 An unsymmetrical transition state requires that radical addition to each carbon of the multiple bond represents a distinct reaction pathway; there is no common intermediate. Also, partial σ‑bond formation between the α-carbon atom and the incoming, carbon-centered radical causes the groups attached to each of these atoms to assume a decidedly pyramidal arrangement; thus, reaction causes the groups attached to each center to move closer together.
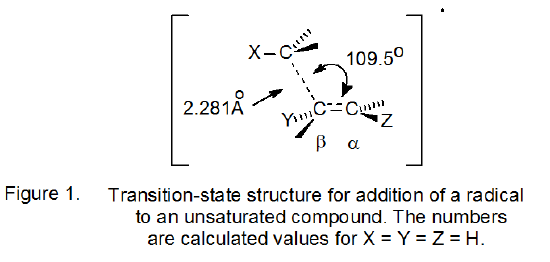
D. Factors Controlling Regioselectivity
The unsymmetrical nature of the transition state structure shown in Figure 1 requires that addition to each carbon atom of an unsymmetrically substituted double bond has a different rate constant for reaction. Understanding regioselectivity in addition reactions then depends upon correctly analyzing the factors controlling these two rate constants. “The temperature dependence of the rate constants is well described by the Arrhenius equation k = Aexp(-Ea /RT). Thus, at a given temperature, the rate variations with radical and substrate substitution can be caused by variations in the frequency factor (A) and/or the activation energy (Ea). For polyatomic radicals, the frequency factors span a narrow range... Hence, the large variation in the rate constants is mainly because of variations in the activation energy”.8 The major factors determining activation energy [bond strengths, steric effects, stereoelectronic effects, and polar effects] are then the ones that need to be considered in determining reaction regioselectivity.8
1. Bond Strengths
A characteristic of many reactions that are similar in nature is that their energies of activation (Ea) can be determined from the Evans–Polanyi relation (eq 4).8,10 (The Evans–Polanyi relation is discussed in Section I.A. of Chapter 7.) In such situations calculating these energies depends upon determining reaction enthalpies (Hr) and establishing values for the two constants in eq 4. For the addition of carbon-centered radicals to C–C double bonds the values for the experimentally determined constants are C=50 kJmol-1 and α=0.25, when Ea and Hr are expressed in kJmol-1.8,15 The number 0.25 for the proportionality constant α means that the enthalpy change, which depends on the difference in the strengths of the bonds being broken and formed, needs to be large for it to have a significant impact on the energy of activation for the reaction. The 0.25 value for α is reasonable for a reaction with an early transition state.
.png?revision=1&size=bestfit&width=140&height=106)
2. Steric Effects
Rehybridization of the β-carbon atom from sp2 to sp3 takes place during radical addition (eq 1). The necessary repositioning of groups that this rehybridization requires forces them closer together (i.e., causes group compression) as reaction proceeds. Any resistance to group compression caused by steric hindrance raises the energy required to reach the transition state for a reaction.8–10 The transition state, therefore, becomes energetically more difficult to attain as the steric size of any of the groups attached to the β‑carbon atom increases. A similar steric compression of the groups attached to the carbon atom bearing the radical center in the adding radical also takes place, but the effect should be smaller because a typical radical center has a structure that already is at an intermediate stage between sp2 and sp3 hybridization.8–10
In addition to group compression, steric interactions at the transition state also arise between groups attached to the β-carbon atom and those bonded to the adding radical (Figure 1). Experimental support for significant interaction comes from the finding that the rate constants for radical addition to the β-carbon atom of an alkene change dramatically when sterically demanding groups are introduced on this atom.7,16 In the reactions represented in eq 5 increasing the steric size of the Y group significantly decreases the rate constant for β addition.7 While it may be difficult to decide how much rate constant reduction is attributable to group compression and how much to interaction between groups on the β-carbon atom and the incoming radical, the relative rate constants shown in eq 5 leave little doubt that steric effects play a major role in determining the rates of radical addition reactions.7
.png?revision=1&size=bestfit&width=415&height=282)
Since the separation at the transition state between the adding radical and the α-carbon atom in an addition reaction is considerable (Figure 1), it is reasonable to expect that any steric hindrance involving α-substituents should be small.7,16 The relative rate constants shown in eq 6 support this expectation because a dramatic change in the steric size of groups attached to the α‑carbon atom has only a small effect on the value of these constants; the largest and the smallest differ only by a factor of 4.2.7
.png?revision=1&size=bestfit&width=415&height=271)
Steric effects have a more important role in determining addition-reaction regioselectivity than do the strengths of the bonds being broken or formed. The reason for this situation can be traced to the nature of the addition process. In the competing reactions that determine regioselectivity [i.e., addition to either the α or β carbon atom in a multiple bond of an unsaturated compound] the same number and types of bonds are being broken and formed; consequently, there should be little difference in activation energies for these two reactions based on bond strengths alone.
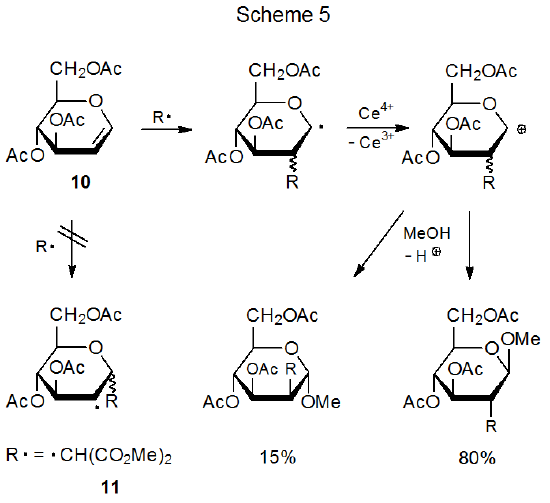
Although the primary role of steric effects in determining regioselectivity in radical addition reactions is clear, these effects are not always the sole determining factor. It would be difficult, for example, to explain preferential addition to C-2 in the glycal 10 (Scheme 5) on the basis of steric effects alone because C-2 is, if anything, more hindered than C-1.17–19 Clearly, another factor also affects regioselectivity in reactions of this type.
3. Polar Effects
Polar effects are influences on reactivity caused by unequal electron distribution within a molecule or reactive intermediate. In radical addition reactions these effects can originate with substituent groups and can be transmitted to the reacting atoms either through bonds or through space. Polar effects also can arise from electron delocalization that produces unequal electron distribution.
The data shown in eq 7 illustrate the importance that polar effects have on radical addition reactions.7 These data describe the relative rate constants for addition of the nucleophilic cyclohexyl radical (C6H11·) to substituted α,β-unsaturated esters. The rate constant is large when a strongly electron-withdrawing substituent (e.g., CN, CO2Me) is attached to the α‑carbon atom in one of these unsaturated esters. Electron withdrawal from the double bond by either CN or CO2Me is due primarily to delocalization that shifts electron density to one of these the α-substituents. In the case where the α‑substituent is a methoxycarbonyl group (Z = CO2Me), the electron-density shift can be seen in the contributing resonance structures shown in Figure 2. Although the polar effects being described are those that exist in the reactants, the rate constants in eq 7 support the idea that these effects remain significant at the transition state.
.png?revision=1&size=bestfit&width=415&height=191)
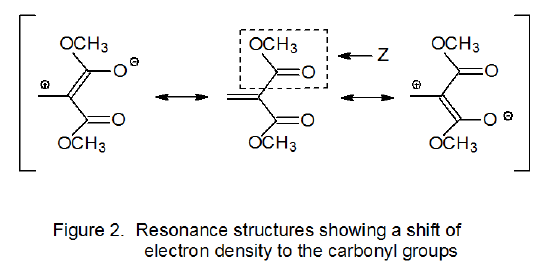
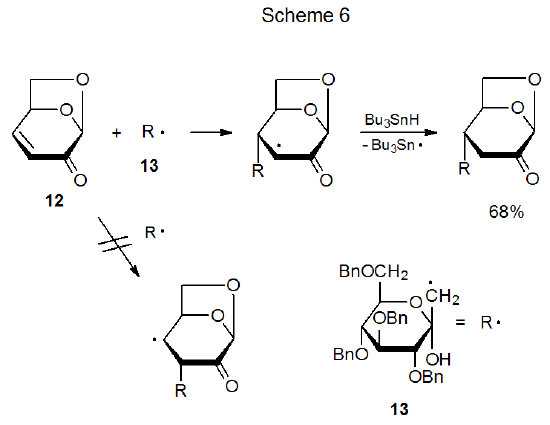
Polar effects not only explain the difference in rate constants for the reactions shown in eq 7 but they also rationalize the regioselectivity of these reactions. The resonance hybrid pictured in Figure 2 indicates a reduced electron density at the β-carbon atom in the carbon–carbon double bond of the ester; consequently, this atom represents a point of attraction for a nucleophilic radical. In such a situation regioselective, β-carbon-atom addition can be expected. An example of this type of addition is shown in Scheme 6 where the nucleophilic carbohydrate radical 13 adds regioselectively to the β-carbon atom of the α,β-unsaturated ketone 12.20
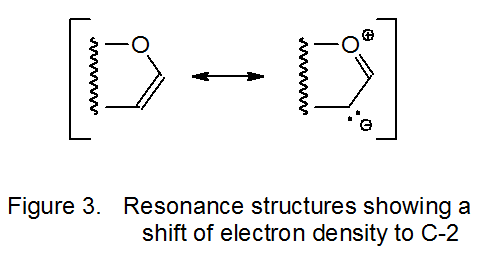
Addition of a nucleophilic radical to an electron-rich double bond is too slow to compete with other radical reactions, but if the radical is electrophilic, addition takes place. The dimethylmalonyl radical 11, for example, adds to the electron-rich double bond in the D-glucal 10 (Scheme 5).17–19 As the resonance hybrid pictured in Figure 3 indicates, C-2 in 10 has greater electron density than C-1; thus, the electrophilic radical 11 not only adds to the double bond in 10 but it does so regioselectivity at C-2 (Scheme 5).
Since steric and polar effects often favor formation of the same product in a radical addition reaction (i.e., that from addition to the least substituted carbon atom in the double bond of the unsaturated reactant), it is sometimes difficult to determine the relative contribution of each effect to the regioselectivity of a reaction. A series of experiments designed to test these contributions is shown in eq 8.7 The first experiment involves addition of the cyclohexyl radical to methyl acrylate (eq 8, R = H). In this reaction both steric and polar effects favor addition of the nucleophilic cyclohexyl radical to the less substituted carbon atom in the carbon–carbon double bond, but as the R group becomes sterically larger, the regioselectivity of the reaction decreases. For the sterically largest R group the favored direction of addition actually changes. The message here is that steric effects can overwhelm polar effects in establishing reaction regioselectivity, but a sterically quite demanding group (e. g., a t-butyl group) is necessary to overcome a strong polar effect.
.png?revision=1&size=bestfit&width=425&height=289)
Another indication of the significance of polar effects in radical addition reactions can be seen by returning to the Evans-Polanyi relation (eq 4). This relation applies to radical addition reactions in which polar factors are not important. For reactions where polar factors are important, energies of activation are lower than those calculated from eq 4. In such situations a modified equation (eq 9), one including the multiplicative terms Fn and Fe, reflecting nucleophilic and electrophilic polar effects, respectively, is more accurate.8,15
.png?revision=1&size=bestfit&width=185&height=163)
4. Frontier-Orbital Interactions
Because radical addition reactions have early transition states,7 frontier-orbital interactions are able to provide an alternative approach for explaining reaction regioselectivity. The first step in this approach is identifying the frontier orbitals in the reaction of interest; for example, in the addition of the dimethylmalonyl radical 11 to the electron-rich double bond in the D-glucal 10 (Scheme 5), the primary interaction is between the SOMO of 11 and the HOMO of 10 (Figure 10 in Chapter 7). Identifying the frontier-orbital interactions in a reaction does not, by itself, explain reaction regioselectivity, but orbital identification is a critical first step for such understanding because from frontier orbitals come the atomic-orbital coefficients that form the basis for explaining regioselectivity.
Atomic orbital coefficients are valuable in determining the regioselectivity of a reaction with an early transition state because the rate constant for the bond-forming reaction between two atoms in such a reaction depends to a large extent on the magnitude of the coefficients in their interacting frontier orbitals.17,18 In the reaction pictured in Scheme 5 the most effective bonding is between the radical 11 and C-2 in the D-glucal 10 because the atomic orbital coefficient at C-2 for the HOMO in 10 is larger than that at C‑1 (Figure 4);21 consequently, regioselective addition to C-2 is favored.17,18
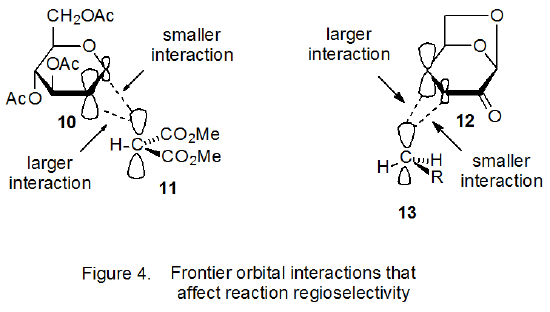
Frontier-orbital interactions also explain regioselectivity in the addition reaction shown in Scheme 6, where a nucleophilic radical (13) is adding to an electron-deficient double bond.20 Addition of the radical 13 to C‑4, rather than C-3, in the α,β-unsaturated ketone 12 cannot be explained by steric effects, but frontier-orbital interactions do provide a basis for understanding the observed regioselectivity. The most important interaction in this case is between the SOMO of 13 and the LUMO of 12. (A justification for this being the primary, frontier-orbital interaction is given in Section IV.B.1 of Chapter 7) For a LUMO such as that in 12 the largest atomic orbital coefficient is associated with the p orbital at C-4 (Figure 4);21 consequently, regioselective addition to C-4 is favored.22
5. Adduct-Radical Stabilization
Adduct-radical stabilization as a possibility for explaining regioselective addition of a carbon-centered radical to a multiple bond is not highly regarded because the exothermic nature of and probable early transition state for radical addition reactions argue against significant, transition-state stabilization due to the developing radical. Evidence from study of model compounds that is cited in support of this point of view is that the cyclohexyl radical adds more rapidly to acrolein and acrylonitrile than to styrene (eq 10),7,23 even though a phenyl group is more effective at stabilizing a radical center than is a carbonyl or cyano group.7,24 This information indicates that adduct-radical stabilization is less important than polar effects at the transition state for an addition reaction; thus, polar effects are primarily responsible for the differences in reactivity of the unsaturated compounds, differences such as those shown in eq 10.7 (As discussed in Section II.D.2 and seen in eq 6, steric hindrance from the α substituents used in the reactions shown in eq 10 should be inconsequential.) Since reactions between electron-deficient alkenes and nucleophilic radicals are stabilized at the transition state by polar effects, these effects could mask less important, adduct-radical stabilization. A better test of the importance of adduct-radical stabilization on regioselective addition would be one in which polar effects could not be the determining factor.
.png?revision=1&size=bestfit&width=380&height=177)
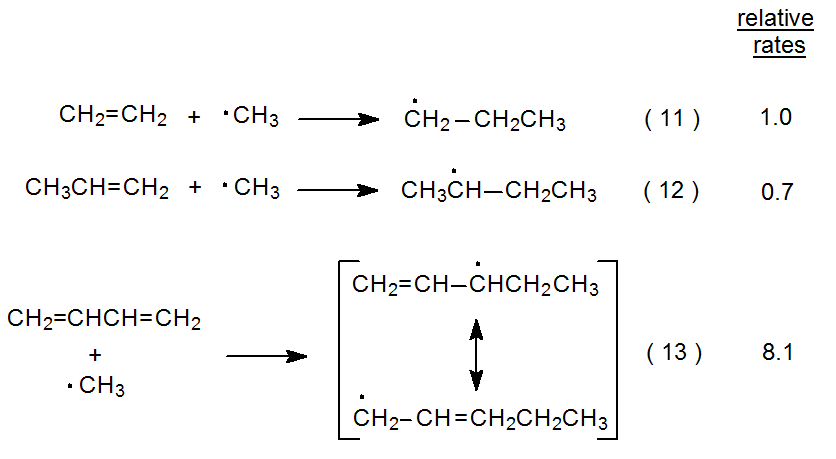
The addition reactions shown in equations 11 and 12 are ones for which polar effects should be minimal.10 The similarity in relative rates for the two reactions indicates that adduct-radical stabilization is inconsequential at the transition state. These reactions also underscore the difficulty in eliminating completely the influence of polar effects when comparing radical reactions. The slightly reduced rate for the reaction shown in eq 12, when compared to that in eq 11, could be due to the effect of the weakly electron-donating methyl group in propene reducing to a small extent the rate of addition of a nucleophilic radical to a slightly more electron-rich double bond. (As mentioned in Chapter 7, Sections III.C. and III.E., there is not complete agreement about the nucleophilicity of the methyl radical.)
-(13).png?revision=1&size=bestfit&width=420&height=231)
The reaction shown in eq 13 supports the idea that the stability of the developing radical can be a factor in reducing transition-state energy.10 The greater rate for this reaction, when compared to those shown in equations 11 and 12, can be explained by resonance stabilization in the developing radical contributing significantly to transition-state stabilization. The limited data in equations 11-13 are consistent with the idea that adduct-radical stability is only a factor in radical addition reactions when such stabilization is considerable. Once again, however, polar effects cloud this interpretation. 1,3-Butadiene can be viewed as a molecule in which each double bond has an ethenyl substituent attached. Such a substituent should be electron-withdrawing or, at least, less electron-donating than a methyl group; consequently, the double bonds in 1,3-butadiene should be more reactive toward the methyl radical than is the double bond in propene. This difference could explain, at least in part, the difference in relative rates for the reactions shown in eq 12 and eq 13.