III. Radical-Center Configuration
- Page ID
- 23931
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)A. Planar and Pyramidal Structures
The configuration of a radical defines the location in space of the atoms directly attached to the radical center. When three such atoms are bonded to the carbon atom upon which a radical is centered, the configuration is either planar or pyramidal.4 A planar configuration is one in which the atoms directly attached to the radical center and the center itself all exist in the same plane (Figure 1). For pyramidal radicals the plane defined by these directly attached atoms no longer includes the central atom (Figure 1).
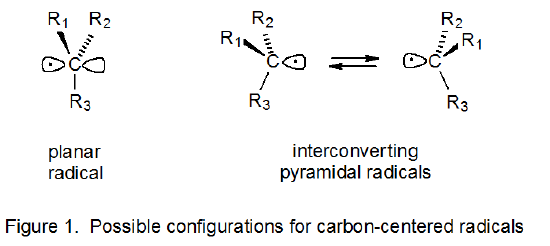
Nearly every carbon-centered radical has a pyramidal configuration at the radical center, but these radicals vary widely in how close their configurations are to being planar.5 A terminology has arisen that is designed to indicate approximate radical configuration. If a radical center has a nearly planar arrangement of attached atoms, the radical is described as being π‑type.6 (Since in a π-type radical the orbital in which the electron is centered is close to being a p orbital, this orbital is often referred to as being p‑type.) If a radical has a decidedly pyramidal configuration (i.e., one approaching that corresponding to sp3 hybridization), the radical is described as being σ‑type.
Pyramidal, carbon-centered radicals with no electronegative substituents attached to the radical center tend to have a small distortion from planarity;5 that is, they usually are considered to be π-type radicals. (It is worth noting that the magnitude of the angle of distortion can be deceptive. The relatively small 6.2o distortion from planarity reported for the ethyl radical means that this radical is actually about 1/3 of the way to being sp3 hybridized.7) The distortion from planar arrangement increases as electron-withdrawing substituents replace other groups attached to the radical center. The change in configuration that accompanies replacement of the hydrogen atoms in the methyl radical by fluorine atoms provides a clear example of the effect of electronegative substituents on radical geometry.5,8,9 The methyl radical is either planar, or nearly so,10 but progressive replacement of hydrogen atoms with fluorine atoms produces pyramidal radicals with structures increasingly further from planarity until the trifluoromethyl radical is reached, in which case the F–C–F bond angles are similar to those found in tetrahedral structures.11
B. Configurational Determination from α-13C Hyperfine Coupling Constants
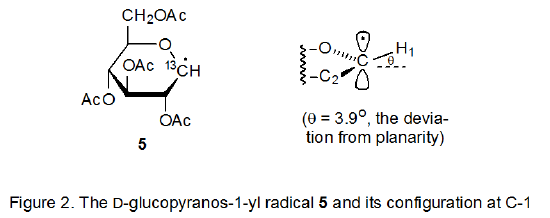
Information about radical configuration can be obtained from analysis of α-13C hyperfine coupling constants. These coupling constants, obtained from the ESR spectra of 13C-enriched radicals, provide a sensitive measure of the hybridization at a radical center.12,13 The configuration of a pyranos-1‑yl radical is naturally of considerable interest due to the unique role of the anomeric carbon atom in carbohydrate chemistry. The α-13C hyperfine coupling constants, obtained from the ESR spectrum of the 13C-enriched D-glucopyranos-1-yl radical 5, show the deviation from planarity for this radical to be 3.9o (Figure 2).6,14 Since an sp3‑hybridized σ radical would have a deviation 19.5o, the D‑glucopyranos-1‑yl radical 5 is considered to be π‑type.6 Radicals centered at C-2 (6), C-3 (7), and C-4 (8) in pyranoid rings also have π‑type configurations.15
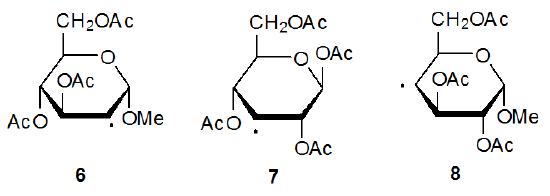
Since organic radicals with no electronegative substituents attached to the radical center have π-type configurations, finding that radicals 6-8 have this type of configuration is not surprising. Because radical centers with electronegative atoms attached become more pyramidal, it is surprising to discover that the radical 5, which has an oxygen atom bonded to the radical center, also has a π-type configuration. To understand why this configuration is adopted, it is helpful to analyze of the stability of 5 as determined by frontier-orbital interactions.
C. Theoretical Explanation of Observed Configurations
1. Frontier-Orbital Interactions
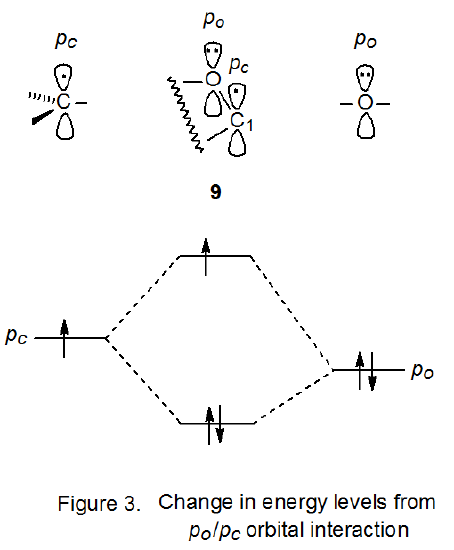
Frontier-orbital interactions are based on an approximate, quantum-mechanical method that assumes that all interactions between occupied orbitals in a bimolecular reaction can be neglected and that the only interactions that need to be considered are between the highest occupied molecular orbital (HOMO) of one reactant and the lowest unoccupied molecular orbital (LUMO) of the other. A small energy difference between the HOMO and the LUMO (the frontier orbitals) translates into a large stabilizing interaction. In radical reactions the singly occupied molecular orbital (SOMO) can be either an HOMO or a LUMO.16 Although frontier-orbital interactions are intended to be applied to bimolecular reactions, they can be used for understanding radical structure. In making such an application the radical is formally split into two fragments and fragment recombination is treated as a bimolecular reaction.16 This approach, which has enjoyed widespread application and success in explaining radical structure,17 will be used to rationalize the π-type configuration at C-1 adopted by the D-glucopyranos-1‑yl radical 5 (Figure 2).
2. pc /po Orbital Interaction
Experimental and theoretical studies show that the two unshared pairs of electrons on an oxygen atom in a pyranoid ring, do not have the same energy.18,19 The higher energy pair exists in a p‑type orbital while the lower energy pair is in a hybrid orbital that has considerable s character. As pictured in Figure 3, stabilization should result from interaction of the electrons in a p-type orbital on a ring oxygen atom (po) with the electron in the singly occupied, p-type orbital on an adjacent carbon atom (pc). The increase in energy of the electron in the singly occupied molecular orbital (SOMO) is more than offset by the combined decrease in energy of the two electrons in the doubly occupied orbital. Because a nonparallel alignment of orbitals would exist in a pyranos-1-yl radical with a σ‑type configuration, stabilizing orbital interaction for such a radical would be less than that for a radical with a π-type configuration, thus, there is a gain in radical stabilization to be had from having a p-type orbital at C-1 even though this atom has an electronegative oxygen atom attached.