
II. Basic Stages of a Radical Chain Reaction
- Page ID
- 23816
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)A. The Initiation Phase
1. Thermal Initiation
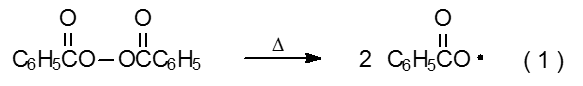
Radical chain reactions usually occur in solutions maintained at 110 oC or less. Most compounds cannot initiate a reaction under these conditions because most molecules do not undergo bond homolysis rapidly enough at or below 110 oC to provide the supply of radicals needed for a productive chain reaction.3 Compounds that can initiate reaction under these conditions often contain a weak σ-bond (e.g., the O–O bond in benzoyl peroxide) that cleaves thermally to generate a pair of radicals (eq 1).
.png?revision=1&size=bestfit&width=305&height=45)
In addition to simple heating of a reaction mixture, thermal initiation also can be brought about sonochemically. Irradiation of homogeneous liquids with high-intensity ultrasound creates localized, superheated, sonochemical cavities in which radicals are generated by thermal reaction.4,5 Sonochemically initiated reactions can be conducted in solutions for which the temperature outside the sonochemical cavities is well below the 80-110 0C that is typical for many radical reactions; for example, sonochemical reaction of tri-n-butyltin hydride in a solution held at 22 oC initiates a chain reaction by homolytically cleaving a tin–hydrogen bond (eq 2).4
.png?revision=1&size=bestfit&width=270&height=37)
An appropriate initiator should provide a steady supply of radicals during the entire reaction. The needed supply will exist if the initiator has a lifetime comparable to the time required for completion of the reaction being conducted.3 [The lifetime of an initiator is often described in terms of the amount of time required for 50% of the material to react, that is, its half-life (t½).] Continuous formation of radicals is necessary because the time of existence of a typical radical chain is short, usually less than one second; consequently, new chains must be started regularly.3 If the half-life of an initiator is too short to provide the necessary supply of radicals during the entire reaction, either an initiator with a longer half-life can be used, or the initiator can be added to the reaction mixture continuously (or at regular intervals) during the reaction.
2. Thermal Initiators
a. 2,2'-Azobis(isobutyronitrile)
2,2'-Azobis(isobutyronitrile) (AIBN) is easily the most widely used initiator in radical reactions of carbohydrates. There are compelling reasons for this status. AIBN has a half-life of one hour at 85 oC (five hours at 70 oC);3,6,7 consequently, it can continuously supply sufficient initiating radicals at moderate temperatures for reactions requiring several hours to reach completion. Other advantages of AIBN are that it is easily handled, generates good yields of radicals (yields of radicals available for chain initiation are not 100% because some radicals combine before they can escape from the solvent cage), and has a rate of decomposition that is almost independent of the solvent.3
The 2‑cyano-2-propyl radical (3) generated from AIBN (eq 3) is relatively stable and usually reacts with bonds that are weaker than most C–H bonds; thus, hydrogen-atom abstraction from the solvent or carbohydrate reactant generally is not a complicating factor. Since the 2-cyano-2-propyl radical readily abstracts a hydrogen atom from a compound with a tin–hydrogen bond, AIBN is an excellent initiator for the frequently encountered reactions in which tri-n-butyltin hydride is the hydrogen-atom source.3
.png?revision=1&size=bestfit&width=400&height=95)
Another advantage of AIBN is that it does not experience induced decomposition; that is, the kinetics of its reaction are first order, regardless of the solvent or initiator concentration.6 (Induced decomposition in this case refers to the more rapid decomposition that sometimes occurs when an initiator undergoes bimolecular reaction in addition to the normal unimolecular one.)
b. 4,4'-Azobis(4-cyanovaleric Acid) (ACBA); 1,1'Azobis-(cyclohexanecarbonitrile) (ABCN); 2,2'-Azobis(2,4-dimethyl-4-methoxyvaleronitrile) (V-70); and Di-tert-butylhyponitrite (TBHN)
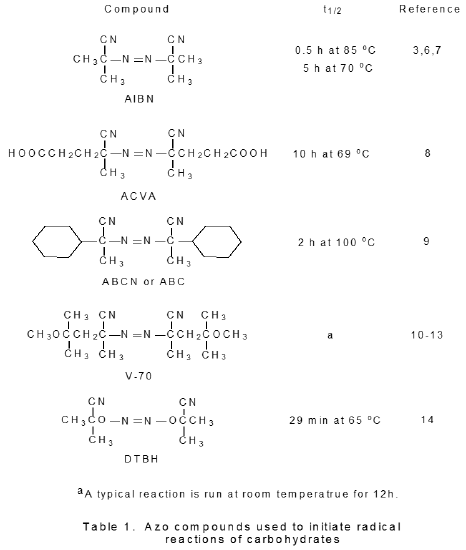
Azo compounds that are known to initiate radical reactions of carbohydrates are shown in Table 1. Even though the vast majority of reactions are initiated by AIBN, each of the other azo compounds listed in Table 1 has a characteristic that is useful in certain situations. The water soluble 4,4'-azobis(4-cyanovaleric acid) (ACBA) initiates reactions run in aqueous solution.15 1,1'-Azobis(cyclohexanecarbonitrile) (ABCN or ABC) has a longer half-life than does AIBN and, thus, is better suited for reactions that require higher temperature or extended reaction times.16,17 2,2'-Azobis(2,4-dimethyl-4-methoxyvaleronitrile) (V-70), in contrast, reacts rapidly enough in solution that it initiates reactions run at or near room termperature.10–13 Di-tert-butyl hyponitrite (TBHN) differs from other azo initiators in that it forms oxygen-centered radicals.14
c. Peroxides
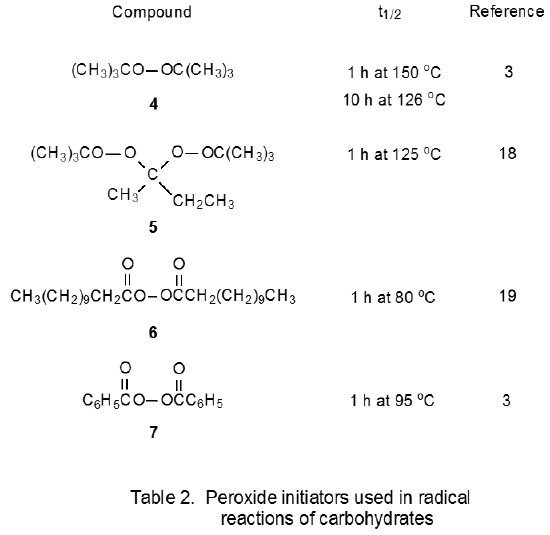
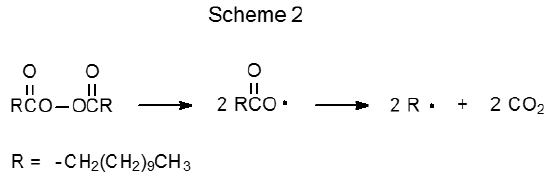
The peroxides shown in Table 2 also are initiators for radical reactions of carbohydrates. These compounds are useful when the 2-cyano-2-propyl radical 3 (eq 3), or any carbon-centered radical generated from other initiators listed in Table 1, does not have the necessary reactivity to cause a chain reaction. Such a situation arises when an initiating radical is required to abstract a hydrogen atom from a carbon–hydrogen bond. The tert-butoxy radicals formed from di-tert-butyl peroxide (4) or 2,2-di-tert-butylperoxybutane18,20–23 (5) are effective at this type of hydrogen-atom abstraction. Sometimes reaction initiation requires a carbon-centered radical that can add to an O-thiocarbonyl group. One radical reactive enough to make this addition is CH3(CH2)9CH2· formed from dilauroyl peroxide (6) (Scheme 2).24–29 Benzoyl peroxide (7) and di-tert-butyl peroxide30–32 (4) also produce radicals that add to an O-thiocarbonyl group.
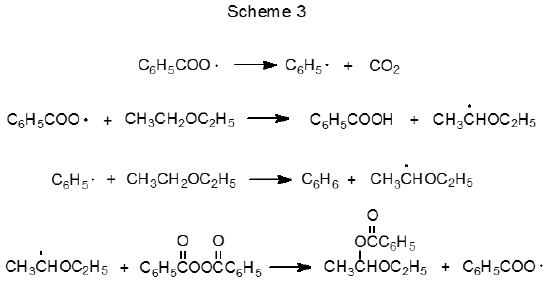
The ability of radicals formed from peroxides to abstract hydrogen atoms from carbon–hydrogen bonds can be both an advantage and a disadvantage. It is an advantage when such abstraction is necessary for reaction to proceed but a disadvantage when hydrogen-atom abstraction from the carbohydrate or the solvent causes undesired reaction. One undesired reaction is induced decomposition, which can be illustrated by considering the reactions of benzoyl peroxide. At low concentrations in an inert solvent the reaction of benzoyl peroxide follows the first-order kinetics expected for unimolecular reaction,6 but at higher concentrations or in the presence of reactive solvents benzoyl peroxide undergoes faster reaction.6,33–35 The half-life of this peroxide in ethyl ether in a sealed tube at 80 oC is five minutes rather than the one hour observed in benzene at 95 oC.6 The enhanced rate of reaction in ethyl ether is attributed to the induced decomposition that occurs when the radical created by abstraction of a hydrogen atom from the solvent by either C6H5· or C6H5CO2· reacts with benzoyl peroxide (Scheme 3).6
3. Photochemical Initiation
Photochemical initiation requires a compound to absorb a photon of light and then use this energy to cleave a bond homolytically and, in so doing, create an initiating radical (or radicals). It is desirable for the initiator to absorb visible or long-wavelength ultraviolet (UV) radiation because being able to absorb this type of light minimizes the possibility that another chromophore in a reactant molecule will be excited and undergo an unwanted photochemical reaction. Photochemical initiation is particularly useful for thermally labile substrates because reaction can be conducted at low temperatures.
Carbohydrates that form radicals as a result of light absorption include iodides, bromides, azides, selenides, hypoiodites, and esters of N-hydroxy-pyridine-2-thione.36 For hypoiodites (eq 4) and esters of N-hydroxypyridine-2-thione (eq 5) photolysis with visible light causes radical formation. Since very few functional groups in carbohydrates react as a result of absorbing visible light, there is little danger that this type of radiation will cause a competing photochemical reaction. For compounds that react with visible light, care must be exercised to protect them from premature reaction due to inadvertent light exposure. Since UV radiation is required for bond breaking in carbohydrate iodides, bromides, azides, and selenides, the possibility for undesired photochemical reaction due to exciting a different chromophore in the substrate is greater when irradiating one of these compounds.
.png?revision=1&size=bestfit&width=220&height=29)
.png?revision=1&size=bestfit&width=290&height=104)
Although radicals form from photolysis of all the compounds mentioned in the previous paragraph, the extent to which a chain reaction takes place depends on both the structure of the substrate and the reaction conditions. In the case of esters of N-hydroxypyridine-2-thione, for example, the bond homolysis shown in eq 5 initiates a chain reaction with a quantum yield that ranges from 6 to 35 when R=(CH2)14CH3 and from 19 to 34 when R=C6H11.37 The range of values for each compound is due to quantum yields being determined under different reaction conditions. (The quantum yield for a reaction is the number of molecules reacted for each photon absorbed; thus, for a radical chain reaction the quantum yield is a measure of the number of propagation cycles produced by each initiating radical.)
Bond homolysis is only one of the possible reactions that can take place when a molecule absorbs a photon of light. Other photochemical reactions (e.g., cycloaddition and geometric isomerization) do not involve radical formation. Reference 36 contains a discussion of both the radical-forming and nonradical-forming photochemical reactions of carbohydrates.
4. Photochemical Initiators
Direct photolysis of carbohydrates is not the only way to initiate their radical reactions photochemically. Irradiation of noncarbohydrates also produces radicals that cause carbohydrate reaction; thus, photolysis of azo compounds, ketones, and hexaalkylditins all generate radicals that initiate chain reactions.
a. 2,2'-Azobis(isobutyronitrile)
2,2'-Azobis(isobutyronitrile) (AIBN) has a maximum absorption at 345 nm in its UV spectrum and fragments when irradiated with light of this wavelength to produce nitrogen and two 2-cyano-2-propyl radicals (eq 3).3,7 Because many compounds are transparent to 345-nm light, it is often possible to generate 2-cyano-2-propyl radicals photochemically in a reaction mixture in which AIBN is the only light-absorbing compound. (In contrast to thermal reaction, photolysis of AIBN has the advantage that it can initiate reactions at or below room temperature.)
b. Acetone and Benzophenone
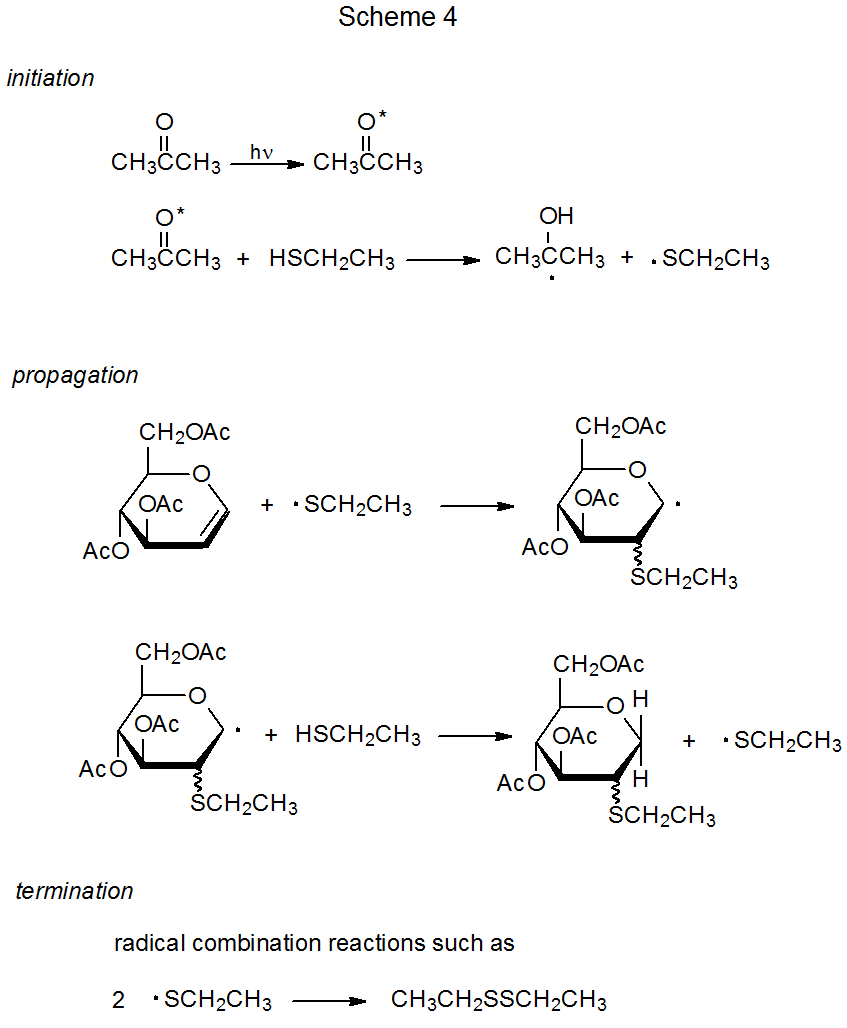
A characteristic of ketones, such as acetone and benzophenone, is that absorption of UV radiation produces an excited state (n,π*) that has considerable radical character on the carbonyl oxygen atom.38 As a result, many excited ketones have reactivity similar to that of alkoxy radicals; in particular, once a photon is absorbed, the excited ketone can abstract a hydrogen atom to begin a chain reaction.38,39 An example of the way in which this type of reaction takes place is shown in Scheme 4, where excited acetone abstracts a hydrogen atom from the S–H bond in ethanethiol to initiate an addition reaction.40
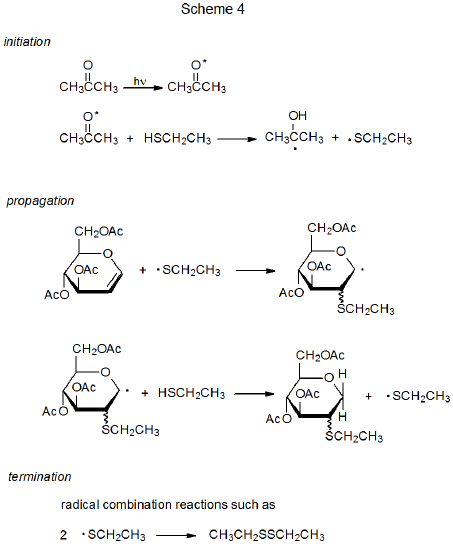
Benzophenone has a longer wavelength and more intense (n,π*) absorption than does acetone; consequently, it is easier to form excited benzophenone without having a carbohydrate reactant absorb the incident light. The presence of the photoproducts benzhydrol and benzpinacol, as well as unreacted benzophenone, can make product purification difficult.41
c. Hexaalkylditins
The chain-carrying radical in many reactions of carbohydrates is the tri-n-butyltin radical (8). This radical usually is generated during the initiation phase of a reaction when the 2‑cyano-2-propyl radical 3 abstracts a hydrogen atom from Bu3SnH (eq 6). Sometimes it is necessary to generate Bu3Sn· without Bu3SnH being present in the reaction mixture. In such a situation Bu3Sn· can be formed by photolysis of hexabutylditin with ultraviolet light (eq 7).42
.png?revision=1&size=bestfit&width=360&height=87)
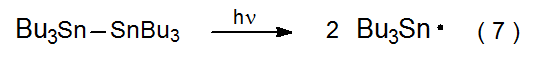.png?revision=1&size=bestfit&width=270&height=30)
5. Chemical Initiation
Thermal and photochemical initiation draw energy for radical formation from heating (vibrational excitation) and photon absorption (electronic excitation), respectively. Chemical initiation is different in that it uses energy stored in bonds to form initiating radicals; for example, triethylboron reacts with molecular oxygen to produce ethyl radicals (eq 8) that then initiate radical reactions.43–46 The driving force behind radical formation is the greater strength of the B–O bond [BDE = 519 kJ/mol (124 kcal/mol) in (EtO)3B] when compared to the B–C bond [BDE = 344 kJ/mol (82.2 kcal/mol) in Et3B].47 The activation energy for this reaction (eq 8) must be exceptionally low because Et3B–O2 can initiate reactions at temperatures as low as - 78 oC.47,48
%252C_(9)%252C_(10).png?revision=1&size=bestfit&width=430&height=154)
Even though ethyl radicals are generated in a reaction involving molecular oxygen (eq 8), they also can react with oxygen molecules to form peroxy radicals (eq 9). Forming peroxy radicals diverts the ethyl radicals from their role as initiators, but this diversion only temporarily interrupts the initiation process because peroxy radicals react with triethylboron to generate new ethyl radicals (eq 10).47 As long as the amount of oxygen added to the system is kept well below the amount of triethylboron present, the ethyl radicals needed to initiate reaction will continue to be produced as oxygen is introduced.
Initiation of a reaction by triethylboron–oxygen shares an important feature with photochemical initiation; namely, both are able to generate radicals well below room temperature. The mild conditions for reactions initiated in this manner can lead to fewer side reactions and higher product yields.49 Triethylboron–oxygen initiation can be used in a broad range of situations that include reaction in aqueous solution.47
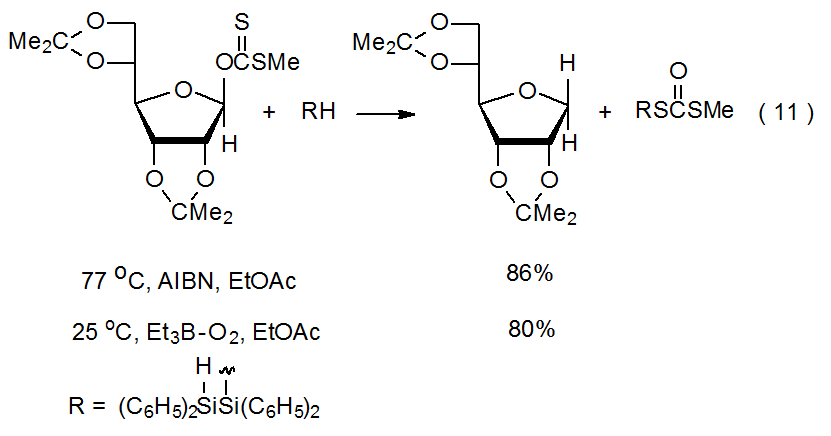
The ability of triethylboron–oxygen to initiate reactions at low temperature indicates that sometimes the sole purpose of heating a reaction mixture is to decompose the initiator. The reaction shown in eq 11, for example, proceeds well at room temperature, when initiated by triethylboron–oxygen. The only apparent reason the reaction must be conducted at higher temperature when AIBN replaces triethylboron–oxygen is to enable fragmentation of the initiator (eq 11).50
.png?revision=1&size=bestfit&width=415&height=218)
The hydrogen-atom donor in most radical reactions is either a tin or silicon hydride. The most frequently used tin hydride is Bu3SnH and the most common silicon hydride is (Me3Si)3SiH. The silicon hydride used in the reaction shown in eq 11 is a less common but effective hydrogen-atom donor.
B. The Propagation Phase
1. General Characteristics
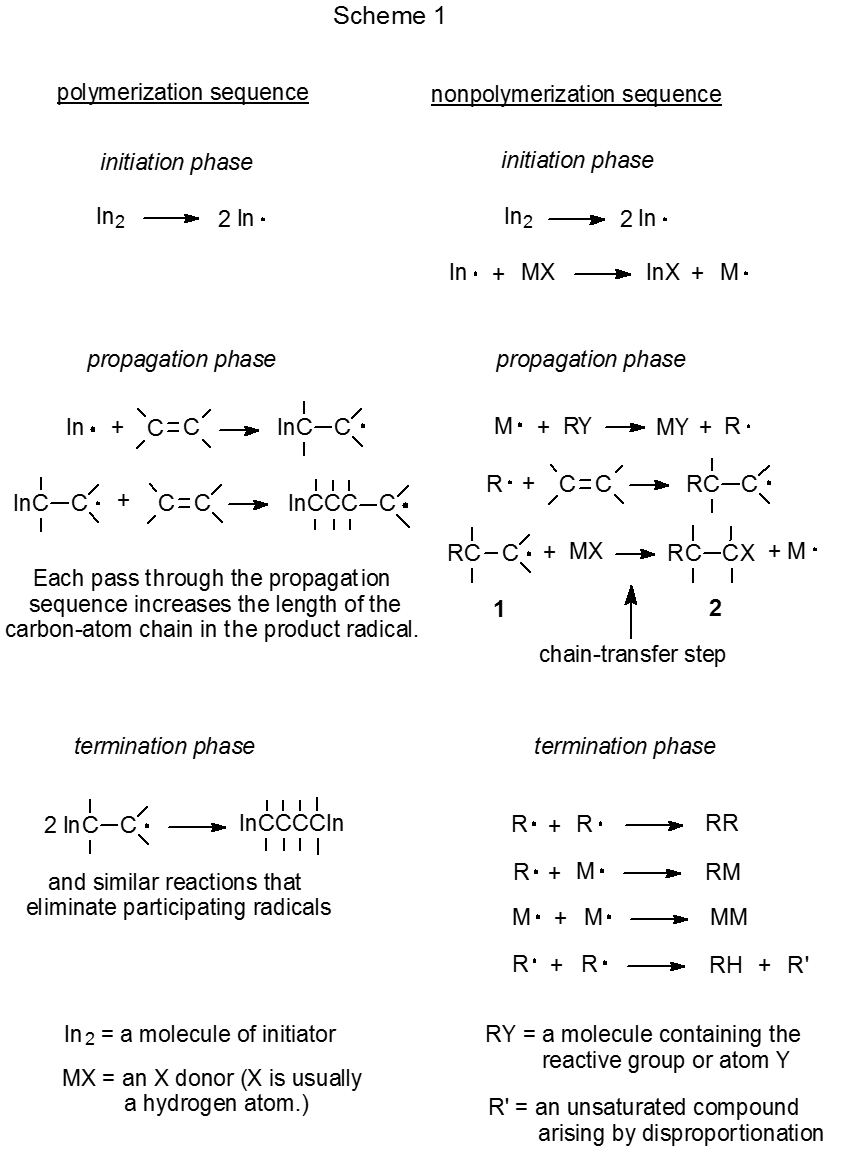
The propagation phase for a chain reaction consists of a group of elementary reactions (also called reaction steps) that are repeated a number of times (Scheme 1). (Elementary reactions are discussed in Chapter 4.) Each repetition represents one reaction cycle, and the number of cycles completed for each initiating radical is the chain length. The final step in a propagation sequence completes the current cycle and generates the radical needed to begin a new cycle. The products from any radical chain reaction are determined by the identity and reactivity of the molecules and radicals participating in the propagation phase.
2. Examples of Propagation Sequences
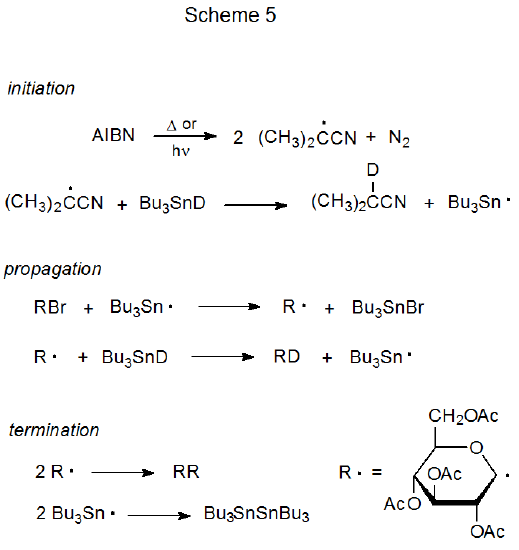
The basic characteristics of a propagation sequence can be seen by examining two specific reactions. One of these is dehalogenation of a D-glucopyranosyl bromide with tri-n-butyltin deuteride51 (Scheme 5), and the other is addition of a D-glucopyranos-1-yl radical to acrylonitrile in the presence of tri-n-butyltin hydride (Scheme 6).52,53

a. Substitution
Reductive dehalogenation with tri-n-butyltin deuteride, a substitution reaction with two propagation steps, is a relatively simple chain process (Scheme 5).51 In the propagation phase for this reaction R· and Bu3Sn· alternate in carrying the chain forward by each reacting only with a particular type of reactant molecule; that is, Bu3Sn· reacts only with the glucosyl halide (RX), and the carbohydrate radical (R·) reacts exclusively with Bu3SnD. This discriminating reactivity has been called "disciplined behavior".54
{kind=link}
b. Addition
Radical addition to an unsaturated compound is a more complex process than atom substitution because radical addition typically involves at least three propagation steps (Scheme 6, desired sequence). Introducing another step (in this case, addition of R· to CH2=CHCN) to the propagation sequence complicates the sequence not just by the existence of a third elementary reaction but also by the constraints this new reaction places on the entire process. These constraints are discussed in the next several paragraphs.
One requirement for success in the addition process shown in Scheme 6 is that R· must be reactive enough to add to acrylonitrile. Carbon-centered, carbohydrate radicals (R·) add rapidly to compounds with electron-deficient multiple bonds but these same radicals add only slowly (too slowly for observable reaction) to compounds with multiple bonds that are not electron-deficient. The reasons behind different rates of addition of R· to compounds with electron-deficient and electron-rich multiple bonds are discussed in Chapter 7. [If an addition reaction is internal (i.e., a radical cyclization), it sometimes will take place even if the multiple bond is not electron-deficient.]
A second requirement for a successful reaction is that R· exist long enough in solution to add to an unsaturated reactant. One reaction that could prevent this addition from taking place is chain termination (R· + R· \(\rightarrow\) RR), but chain termination is unlikely to do so because the very low concentration of R· causes the rate of radical combination to be much slower than the rate of addition of R· to a compound with an electron-deficient multiple bond. A more probable reason for R· not adding to an unsaturated compound is that this radical undergoes hydrogen-atom abstraction from tri-n-butyltin hydride before addition can occur (Scheme 6, diverting sequence). Such an abstraction diverts reaction away from the desired product. The rate of addition of R· to an unsaturated compound, therefore, must be faster than its rate of hydrogen-atom abstraction from tri-n-butyltin hydride in order to keep the chain reaction from being directed into an unwanted propagation sequence (Scheme 6).
A third constraint placed on the overall addition process by the reaction of R· with acrylonitrile is that the adduct radical must not add to a second molecule of the nitrile before hydrogen-atom abstraction takes place. Although this competing addition sometimes does occur, in most instances hydrogen-atom abstraction is more rapid than a second radical addition.
Another way of describing the constraints placed on the desired sequence in Scheme 6 by an additional propagation step is in terms of chain-transfer reactions. In each propagation sequence the chain-transfer step ends the existing cycle and creates the radical that begins a new cycle. In the reactions shown in Scheme 6 the third step in the desired sequence and the second step in the diverting sequence are the chain-transfer steps for their respective reactions. Radical addition cannot be successful if the rate of the chain-transfer step in the diverting sequence is greater than the rate of the radical-transforming step (i.e., addition of R· to CH2=CHCN) in the desired sequence. In other words, the addition reaction shown in Scheme 6 will be a minor process if most of the time R· reacts with tri-n-butyltin hydride to produce RH (the simple-reduction product) before it adds to acrylonitrile.
3. Rate Constants and Reaction Rates
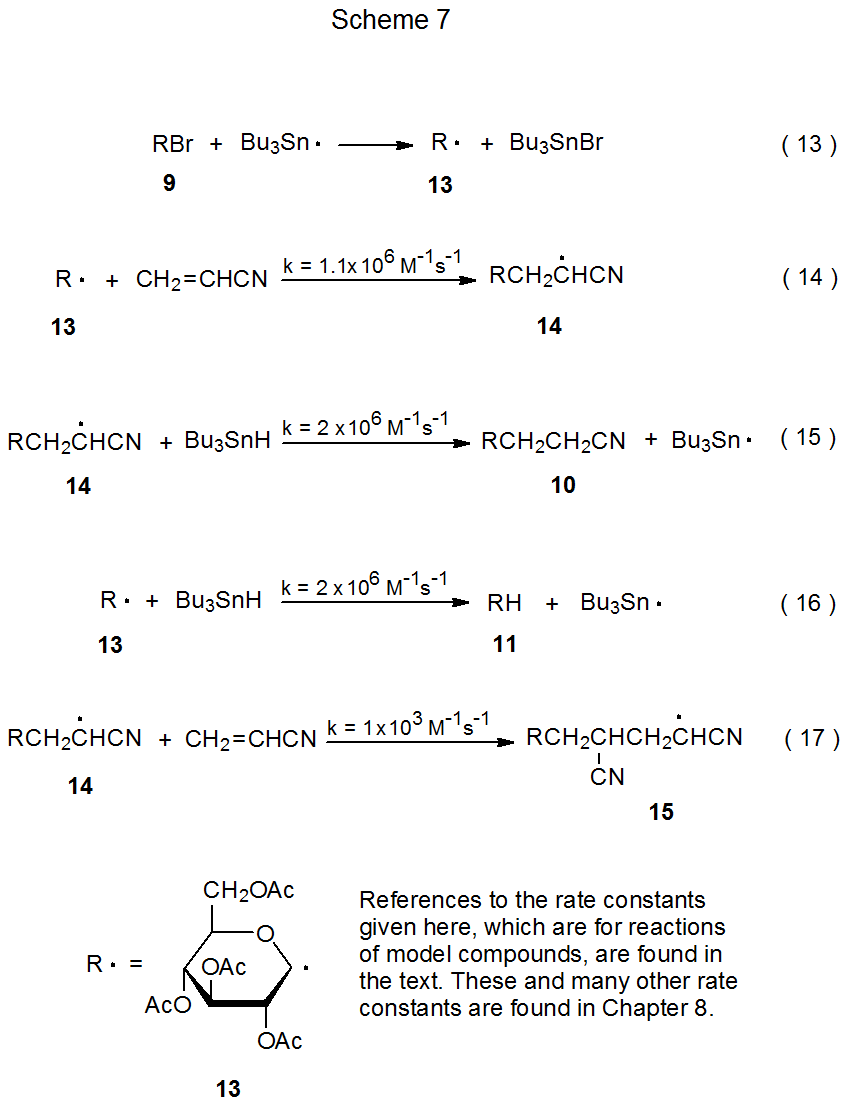
Analysis of the reaction shown in Scheme 6 can be done in a more quantitative manner using the rate constants shown in Scheme 7 and the information given in eq 12.52 (The rate constants in Scheme 7 are a few of the many listed in the tables located in Chapter 8.) The first step in the formation of any product in this reaction is bromine-atom abstraction by the tri-n-butyltin radical (eq 13). (Equations 13-17 are found in Scheme 7.) After bromine-atom abstraction, the major reaction product (10) is formed by a combination of two steps, addition of the D-glucopyranos-1-yl radical 13 to acrylonitrile to give the adduct radical 14 (eq 14) and abstraction by 14 of a hydrogen atom from tri-n-butyltin hydride (eq 15). These reaction steps (equations 14 and 15) need to take place in preference to those that produce the side products 11 and 12 (eq 12). Which product will be the major one and which will be the minor ones in this reaction is determined by reactant concentrations and rate constants for the various, possible elementary reactions.
.png?revision=1)
-(17).png?revision=2)
Unfortunately, not all of the rate constant information needed to analyze the reaction shown in eq 12 is available; consequently, in order to proceed it is necessary to estimate some rate constants based upon information from reaction of model radicals. For example, the rate constant for addition of the pyranos-1-yl radical 13 to acrylonitrile (eq 14) is assumed to be similar to that for addition of the model radical ·CH2OH to this nitrile.55 The rate constant for this reaction (eq 14) is smaller than that for hydrogen-atom abstraction by a typical carbon-centered radical from tri-n-butyltin hydride (TBTH) (eq 16);56 therefore, if acrylonitrile and TBTH are present in equal amounts, the major reaction product should be compound 11. The desired reaction (eq 14) will be favored, however, if acrylonitrile is present in much greater amount than TBTH. In the reaction shown in eq 12 the ratio of acrylonitrile to TBTH (5/1) is large enough to make 10 the major product (58%).52 The 21% yield of 11, however, shows that even with an excess of acrylonitrile there still is substantial reaction of the radical 13 with tri-n-butyltin hydride (eq 16).
There is a limit to how large the ratio of acrylonitrile to tri-n-butyltin hydride can become before adding more nitrile to the reaction mixture is counterproductive. This limit will be reached when reaction of 14 with acrylonitrile (eq 17, k = 1 x 103 M‑1 s-1 )57 competes effectively with reaction of this radical (14) with TBTH (eq 15, k = 2 x 106 M-1 s-1).56 The difference in rate constants for these two reactions is so great, however, that having acrylonitrile present in fivefold excess is possible without significant formation of the radical 15 (eq 17) and its hydrogen-atom abstraction product 12 (eq 12). The considerable difference in the rate constants for the reactions shown in eq 14 and eq 17 is due primarily to a difference in radical philicity; that is, 13 is more nucleophilic than 14. Radical philicity and its effect on radical reactivity are discussed in Chapter 7.
4. Solvent Effects
Although solvent selection often plays a decisive role in ionic reactions, strong solvent effects in radical reactions are less common because radical centers are relatively nonpolar and, thus, are not highly solvated.3 A similar statement applies to transition states in radical reactions because most reactions do not develop much, if any, separation of charge at the transition state; hence, stabilization by polar solvents is usually not a significant factor.
There are some situations in which solvent effects are significant in radical reactions.58 One of these occurs during phosphatoxy group migration.59 The rate determining step in this reaction involves formation of a polarized transition state on its way to becoming an ion pair (Scheme 8); consequently, reaction is faster in solvents that stabilize charge separation. Acyloxy and phosphatoxy group migrations are similar in many ways, including more rapid reaction in polar solvents.

C. The Termination Phase
Any process that stops the participation of a radical in a propagation sequence terminates the reaction chain. In typical chain reactions, such as those described in Schemes 1, 4, 5, and 6, radical combination and disproportionation are terminating steps. Chain termination in this way is limited primarily by how rapidly radicals diffuse through a solution.60,61 Successful chain reactions have concentrations of radicals low enough (approximately 1 x 10-7 M)57,62 to prevent even diffusion controlled processes from competing effectively with chain propagation. Because low radical concentrations make chain-terminating reactions less competitive, adding only a limited amount of an initiator with an appropriate half-life to a reaction mixture generates the small but steady supply of radicals required to maintain reaction and avoid premature chain termination.
{kind=link}
{kind=link}
Chain termination can occur when a participating radical is transformed into a radical that is not involved in the reaction sequence.63 A common way for this to happen is to have a carbon-centered radical (R·) combine with a molecule of oxygen to give a peroxy radical (ROO·) (eq 18). Formation of ROO· would terminate propagation sequences such as those shown in Schemes 5 and 6 because a peroxy radical is not a participant in either sequence. (Forming a peroxy radical by reaction with oxygen not only terminates a desired propagation sequence, but it also creates a reactive radical that is capable of becoming part of a different reaction sequence.) Because reaction of carbon-centered radicals with molecular oxygen occurs at or near diffusion control (k = 109-1010 M-1s-1),64 minimizing chain termination by oxygen, requires radical reactions to be conducted in an inert atmosphere such as that provided by argon or nitrogen.
.png?revision=1)
Even when a reaction is conducted in an inert atmosphere, it is nearly impossible to eliminate all traces of oxygen from a reaction mixture. A small amount of oxygen can cause initial reaction chains to have short lengths. Once the oxygen concentration falls to a low enough level that premature chain termination is no longer significant, reaction can proceed without noticeable inhibition by oxygen.