
6.3: IR Spectrum and Characteristic Absorption Bands
- Page ID
- 359595

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)35
6.3 IR Spectrum and Characteristic Absorption Bands
Xin Liu
With the basic understanding of the IR theory, we will take a look at the actual output from IR spectroscopy experiments, and learn how to get structural information from IR spectrum. Below is the IR spectrum for 2-hexanone.
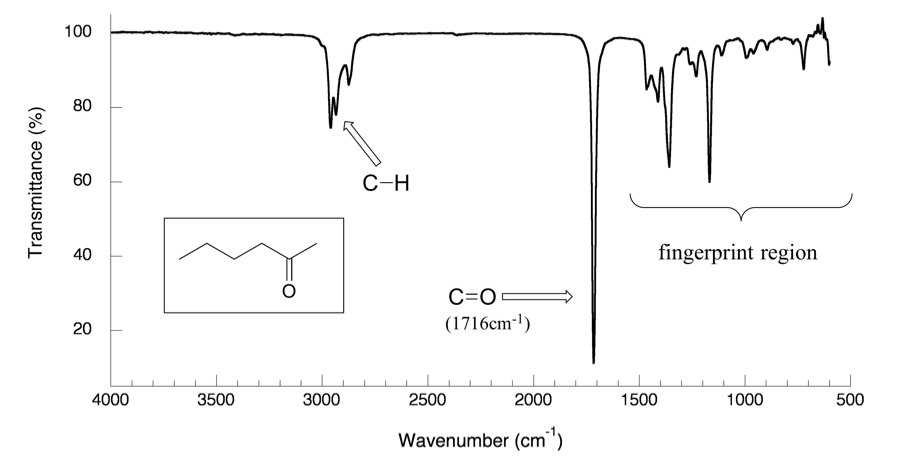
Notes for interpreting IR spectra:
- The vertical axis is ‘% transmittance’, which tells how strongly light was absorbed at each frequency. The solid line traces the values of % transmittance for every wavelength passed through the sample. At the high end of the axis, 100% transmittance means no absorption occurred at that frequency. Lower values of % transmittance mean that some of the energy is absorbed by the compound, and gives the downward spikes. The spikes are called absorption bands in an IR spectrum. A molecule have a variety of covalent bonds, and each bond have different vibration modes, so the IR spectrum of a compound usually show multiple absorption bands.
- The horizontal axis indicates the position of an absorption band. But instead of using frequency to show the absorbed radiation, wavenumbers (
 , in unit of cm-1) are used as a conventional way in IR spectra. The wavenumber is defined as the reciprocal of wavelength (Formula 6.3), and the wavenumbers of infrared radiation are normally in the range of 4000 cm-1 to 600 cm-1 (approximate corresponds the wavelength range of 2.5 μm to 17 μm of IR radiation).
, in unit of cm-1) are used as a conventional way in IR spectra. The wavenumber is defined as the reciprocal of wavelength (Formula 6.3), and the wavenumbers of infrared radiation are normally in the range of 4000 cm-1 to 600 cm-1 (approximate corresponds the wavelength range of 2.5 μm to 17 μm of IR radiation).
Please note the direction of the horizontal axis (wavenumber) in IR spectra decrease from left to right. The larger wavenumbers (shorter wavelengths) are associated with higher frequencies and higher energy.
The power of infrared spectroscopy arises from the observation that the covalent bonds characterizing different functional groups have different characteristic absorption frequencies (in wavenumber, Table 6.1). The technique is therefore very useful as a means of identifying which functional groups are present in a molecule of interest.For example, the most characteristics absorption band in the spectrum of 2-hexanone (Figure 6.3a) is that from the stretching vibration of carbonyl double bond C=O, at 1716 cm-1. It is a very strong band comparing to the others on the spectrum. A strong absorbance band in the 1650-1750 cm-1 region indicate that a carbonyl group (C=O) is present. Within that range, carboxylic acids, esters, ketones and aldehydes tend to absorb in the higher wavenumber/frequency end (1700-1750 cm-1), while conjugated unsaturated ketones and amides tend to absorb on the lower wavenumber/frequency end (1650-1700 cm-1).
Stretching Vibrations
Generally, stretching vibrations the stretching vibrations require more energy and show absorption bands in the higher wavenumber/frequency region. The characteristics stretching vibration bands associated with the bonds in some common functional groups are summarized in Table 6.1.
| Formula | Bond | Characteristic IR Frequency range (cm-1) |
| alcohol | O-H stretching | 3200 – 3600 (broad) |
C=O stretching
C-H stretching
C=O stretching
O-H stretching
C=C stretching
vinyl =C-H stretching
C=C stretching
C≡C stretching
terminal ≡C-H stretching
C-H stretching
N-H stretching
Table 6.1 Characteristic IR Frequencies of Stretching Vibrations
The information in Table 6.1 can be summarized in the diagram that is easier to be identified (Figure 6.3b), in which the IR spectrum is divided in several regions, with the characteristic band of certain groups labelled.
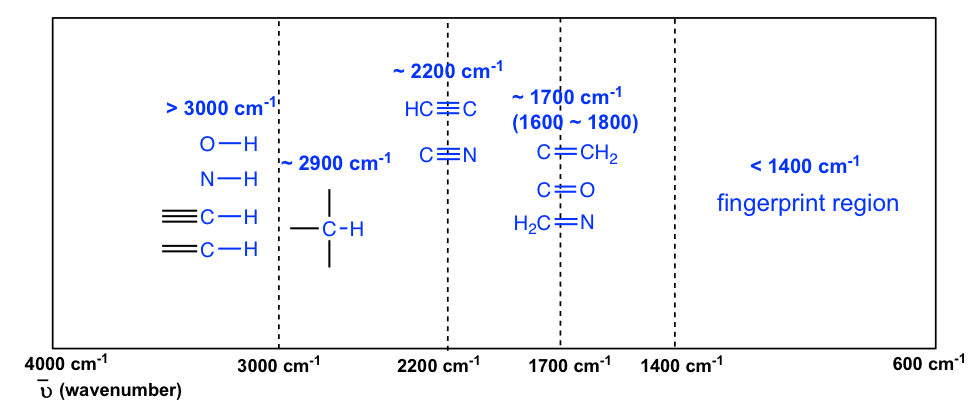
The absorption bands in IR spectra have different intensity, that can usually be referred to as strong (s), medium (m), weak (w), broad and sharp. The intensity of a absorption band depends on the polarity of the bond, the bond with higher polarity will show more intense absorption band. The intensity also depends on the number of bonds responsible for the absorption, the absorption band with more bonds involved has higher intensity.
The characteristic IR frequencies of stretching vibrations in Table 6.1 and Figure 6.3b provide very useful information to identify the presence of certain functional group, that can be generally summarized as:The polar O-H bond (in alcohol and carboxylic acid) usually shows strong and broad absorption bands that are easy to be identified. The broad shape of the absorption band results from the hydrogen bonding of the OH groups between molecules. The OH bond of alcohol group usually has absorption in the range of 3200-3600 cm-1, while the OH bond of carboxylic acid group occurs at about 2500-3300 cm-1 (Figure 6.4a and Figure 6.4c).
The polarity of N-H bond (in amine and amide) is weaker than OH bond, so the absorption band of N-H is not as intense, nor that broad as O-H, and the position is in 3300-3500 cm-1 region.
The C-H bond stretching of all hydrocarbons occur in the range of 2800-3300 cm-1, and the exact location can be used to distinguish between alkane, alkene and alkyne. Specifically:
- ≡C-H (sp C-H) bond of terminal alkyne give absorption at about 3300 cm-1
- =C-H (sp2 C-H) bond of alkene give absorption at about 3000-3100 cm-1
- -C-H (sp3 C-H) bond of alkane give absorption at about ~2900 cm-1 (see the example of IR spectrum of 2-hexanone in Figure 6.3a, the C-H absorption band at about 2900 cm-1)
A special note should be taken for the C-H bond stretching of an aldehyde group that shows two absorption bands, one at ~2800 cm-1 and the other at ~ 2700 cm-1. It is therefore relative easy to identify the aldehyde group (together with the C=O stretching at about 1700 cm-1) since essentially no other absorptions occur at these wavenumbers (see the example of IR spectrum of butanal in Figure 6.4d ).
The stretching vibration of triple bonds C≡C and C≡N have absorption bands of about 2100~2200 cm-1. The band intensity are in medium to weak level. The alkynes can generally be identified with the characteristic weak but sharp IR absorbance bands in the range of 2100-2250 cm-1 due to stretching of the C≡C triple bond, and terminal alkynes can be identified by their absorbance at about 3300 cm-1, due to stretching of sp C-H.
As mentioned earlier, the C=O stretching has strong absorption band in the 1650-1750 cm-1 region. Other double bonds like C=C and C=N have absorptions in bit lower frequency regions of about 1550-1650 cm-1. The C=C stretching of an alkene only shows one band at ~1600 cm-1 (Figure 6.4b), while a benzene ring is indicated by two sharp absorption bands, one at ~1600 cm-1 and one at 1500-1430 cm-1 (see the example of IR spectrum of ethyl benzene in Figure 6.4e).
You will notice in Figure 6.3a and 6.3b that a region with the lower frequency 400-1400 cm-1 in the IR spectrum is called the fingerprint region. Kind of like a human fingerprint, the pattern of absorbance bands in the fingerprint region is characteristic of the compound as a whole. Even if two different molecules have the same functional groups, their IR spectra will not be identical and such difference will be reflected in the bands in the fingerprint region. Therefore the IR from an unknown sample can be compared to a database of IR spectra of known standards in order to confirm the identification of the unknown sample.