
4.6: Interpretation
- Page ID
- 432183
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)- After completing this section, you should be able to
- be able to read an IR spectrum
- understand where the different regions for bonds in the IR spectrum are
- determine what type of hydrocarbon you have in your molecule
When analyzing IR spectra, there are a few things to remember:
1. You are looking to determine what functional groups are a part of a molecule. This is not a method for complete elucidation of a structure for the molecule.
2. You do NOT need to analyze every single peak. For example, the fingerprint region (1500 - 600 cm-1) is a forest of peaks, so this region is often ignored in analysis.
3. Remember, similar functional groups will have similar frequencies, which also indicates certain bonds will fall in the same region.
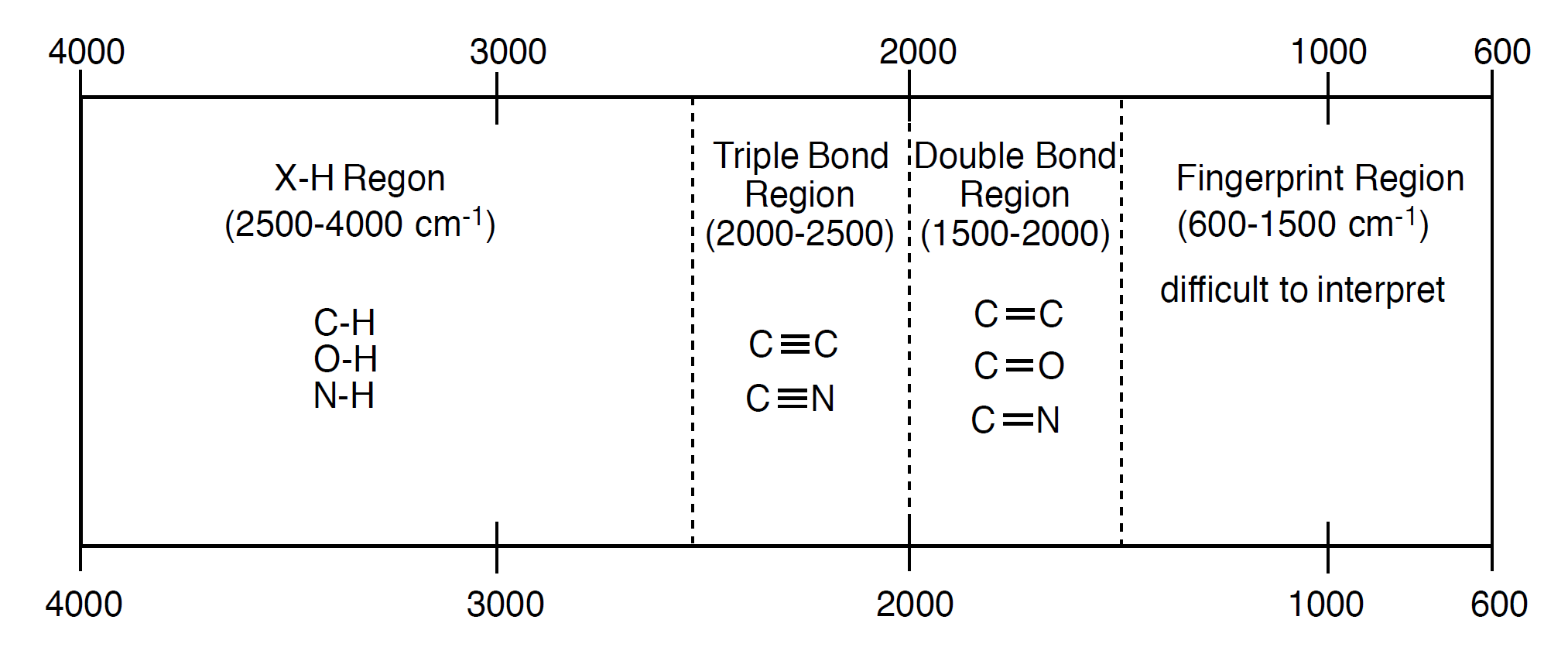
From this, different regions have been determined by previous research. The hydrogen bond region falls from 4000 to 2500 cm-1. This is the area you will find all of your O-H, N-H, and C-H bonds that typically appear in organic molecules. The next region is the triple bond region (2500 - 2000 cm-1), where the C≡C and C≡N bonds commonly found in organic molecules will absorb. The triple bond region leads into the double bond region (2000 - 1500 cm-1). Here you will find the C=C, C=O, and C=N bonds. Finally, the fingerprint region is 1500 - 600 cm-1, where all the single bonds will be found with the exception of some of the bonds to hydrogen that are found in the hydrogen bond region. Below is an image that summarizes what was just described. It is very helpful to have an idea of where these regions are when interpreting an IR spectrum, even with the data table in hand.
In alkanes, which have very few bands, each band in the spectrum can be assigned:
- Let's take a deeper look at pentane. Since most organic compounds have these features, these C-H vibrations are usually not noted when interpreting a routine IR spectrum, which appear at 2873 - 2958 cm-1 in this spectrum. Note that the change in dipole moment with respect to distance for the C-H stretching is greater than that for others shown, which is why the C-H stretch band is the more intense. The peak at 1460 cm-1, is due to C-H scissoring. 1379 cm-1 is a methyl rock. The C-H scissoring and methyl rock an often get lost in the fingerprint region when looking at more complex molecules. Since so many molecules with have similar peaks to an alkane, we can consider these our baseline. Therefore, the regions that will be of interest will be the hydrogen bond region, triple bond region, and double bond region when determining what functional groups are present or if there are other characteristic functional groups present.

In spectrum of 1-hexene, a terminal alkene, is shown below. Here will will look for differences compared to pentane. For alkenes, there will be a Csp2-H peak in the hydrogen bond region (if there are hydrogens attached to the double bond carbons as in this case) and the C=C bond. For 1-hexene, the Csp2-H peak is at 3079 cm-1 and the C=C bond is at 1642 cm-1. At 992 cm-1 and 907 cm-1, there are peaks due to the =C-H bend, but often get lost in the fingerprint region, so will not be a notable peak.

The spectrum of 1-hexyne, a terminal alkyne, is shown below. The notable peaks for the alkyne are the peak greater than 3000 cm-1 and the peak in the triple bond region. In 1-hexyne, there is a peak at 3309 cm-1, which is due to the Csp-H bond. The other peak of note is at 2118 cm-1 for the C≡C. We can ignore the peaks at 2873 cm-1 to 2959 cm-1 because these are your Csp3-H bonds, which are in so many organic molecules that they are not distinctive to the triple bond. This goes for the fingerprint region peaks as well.

Here, we have looked at hydrocarbons, but these are not the only functional groups to consider in organic molecules. In the next section, the focus will be on functional groups containing a heteroatom.
What regions of the IR would you focus on to determine if you have an alkane or alkene?
- Answer
-
If we use the alkane as our baseline, the regions that would stick out for an alkene are the hydrogen bond region above 3000 cm-1 and the double bond region.
What two regions do you find the alkyne notable peaks?
- Answer
-
The hydrogen bond region for the Csp-H bond and the triple bond region for the carbon-carbon triple bond.
Below is a spectrum of toluene. How does it differ from a plain alkene like 1-hexene?

- Answer
-
Both still have a peak around 3000 cm-1 for the Csp2-H bond. However, due to conjugation the C=C shifts to a lower wavenumber in an aromatic molecule. It shifts to about 1600 cm-1 due to conjugation.