
5.3: Tetracyclines
- Page ID
- 285458

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Acetyl CoA is not the only thioester that may initiate the enzyme matrix oligomerization of malonyl CoA. Thus, for example, a biosynthesis has been postulated for tetracycline (44) involving condensation of malonamoyl CoA (34) with eight molecules of malonyl CoA to produce an enzyme bound polyketoamide thioester 35, that is partially dehydrocyclized after methylation of one methylene and reduction of one carbonyl.6 The final ring of the tetracycline ring system is formed by Dieckmann cyclization of 36 to 37 after release of the partially cyclized polyketide from the polyketide synthetase. Two aromatic hydroxylations increase the functionality after completion of the carbon skeleton. These hydroxylations may involve intermediate arene oxides, e.g. 37 → 38 → 39. The reductive amination of 40 via 41 and 42 to yield 43 is accompanied by oxidative deamination of glutamic acid.
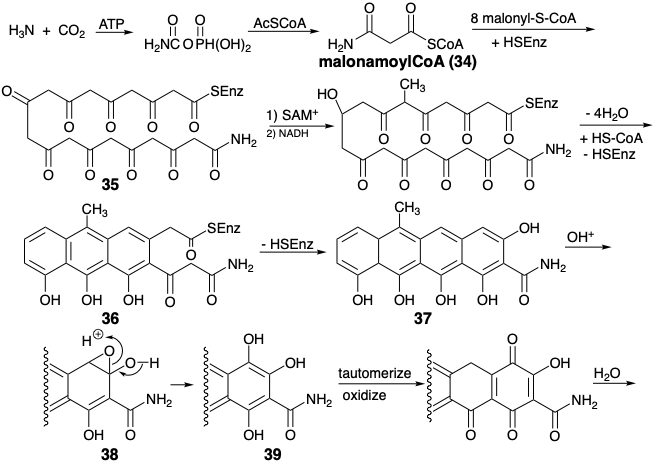

Topological Analysis of Fused-Ring Systems.7
A ring pair is a fused-ring system if two rings share one and only one common bond, the fusion bond. The steroid and tetracycline ring systems are examples of multicyclic structures containing only fused-ring pairs, as opposed to spirocyclic or bridged-ring pairs. Besides fusion bonds (marked f), the diagrams below indicate common atoms (shown as •), and exendo bonds (marked e) which are exo to one ring and endo to another, and bonds (indicated as dashed lines) that are formed during the biosyntheses of these ring systems from acyclic precursors.

There is an interesting contrast between the topological strategies of tetracycline and steroid biosyntheses. Both strategies involve key acyclic intermediates that incorporate all of the skeletal carbon atoms. However, the biosynthesis of the tetracycline skeleton involves formation of all of the ring fusion bonds, whereas only exendo bonds are formed during the biosynthesis of the steroid skeleton. Another contrast is found in the biosynthetic generation of the peripheral ring, i. e. the ring that remains after cleavage of all fusion bonds. Thus, only one peripheral bond is generated during the biosynthesis of the tetracycline skeleton, whereas four bonds of the peripheral ring are generated during steroid biosynthesis.
Also noteworthy is the fact that, in both biosyntheses, bonds between pairs of common atoms are strategic bond, i.e., strategically important in achieving rapid reduction of molecular complexity by disconnection during dislocation of the synthetic target. The dislocations of the tetracycline biosynthetic strategy are recommended both by topological and polar reactivity analysis. Topologically, the strategy disconnects all bonds between pairs of common atoms. Polar reactivity analysis reveals ample functionality. If the activation provided by several functional groups is ignored, numerous functional groups remain with solely consonant connecting circuits that can be generated by exploiting target related functionality in precursors in conjunction with several added consonant functional (carbonyl) groups.

A Linear Strategy for Tetracyclines
The first synthesis of a biologically active (though unnatural) tetracycline derivative was achieved by Woodward and collaborators.8 These workers simplified the synthetic goal by not including the labile tertiary 6-hydroxyl as well as the 6-methyl of tetracycline (44). Ring A of 45 is functionally and stereochemically the most complex portion of this simplified target. This ring is so highly functionalized that polar analysis is ambiguous. It contains a plethora of polar reactivity dissonances. If this ring is severed from 45, a tremendous simplification of the synthetic target results. Not only is an abundance of reactive functionality removed, but a topological simplification is also realized. Thus, by cleaving a pair of exendo bonds that are vicinal and cocyclic (in the same primary ring, i. e. one that is not disected into a pair of smaller rings by a transanular bridge), all vestiges of the A-ring are removed. The remaining BCD synthon is a relatively chemically stable, structurally simple fragment 46. A possible synthetic intermediate, i. e. appropriately functionalized molecular fragment, that corresponds to 46 is 47. The carbonyl group in ring B in 47 provides activation for elaboration of ring A. The methyl ether in ring D blocks deprotonation of the phenol. Polar analysis of the nonaromatic portion of 47 suggests a dislocation to 48 and dimethyloxalate, a symmetrical dissonant biselectrophile. Annelation of 48 by Friedel-Crafts acylation of 49 would exploit the nucleophilic reactivity of the aromatic D-ring in 49, that is activated by a target-related methoxy group, and the electrophilic reactivity of a carbonyl.

However, this strategy is potentially flawed because electrophilic substitution that must occur ortho to the methoxy substituent in the D ring of 49 during conversion to 48, could also occur at the nucleophilic position para to the activating methoxy group. To preclude para acylation, a chloro substituent in the precursor 50 could be used as a blocking group (a substituent introduced to control reactivity and subsequently removed).

Topologically, a dislocation of 49 to 51 + 52 is desireable because all vestiges of the sidechain are removed from the aromatic ring. However, polar analysis of 49 shows that electrophilic alkylation or acylation of an anisole precursor would favor ortho or para substitution rather than the meta substitution required to generate 49.

The dislocation 49 ⇒ 53 + 54 is recommended by the ready availability of benzylic organometallics corresponding to 53. A nucleophilic synthon such as 53 might afford a diester of 49 by 1,4-addition to 54. In fact, a general synthesis of β-substituted alkanedioic acids such as 49 is known that is related to this approach.9 Dislocation to more connected, cyclic β-ketoester intermediates 55 reveals the possible utility of readily available cycloalkenone precursors for the synthesis of β-substituted alkanedioic acids by retro Dieckman cleavage.

In practice, C-acylation of enolate intermediates such as 56 is accompanied by O-acylation of the resulting β-keto esters. However, no additional steps are required because the resulting enol esters are hydrolyzed to β-keto esters under the reaction conditions required for retro Dieckman cleavage of the latter to generate the target β-substituted alkane dioic acids.

Instead of a benzylic nucleophile as starting material, Woodward's strategy was based on the choice of a readily available benzylic electrophile, methyl m-anisate (57), as starting material. This choice channels retrosynthetic analysis to a precursor 58 with an activating functional group, the benzylic carbonyl, that must be removed subsequently to provide 49. Although conjugation with the remote carbomethoxyl in 58 could provide the nucleophilic activation required to unite a hexanedioic ester starting material with 57, Woodward chose to exploit a classical synthetic method for ketones, alkylation of a β-keto ester followed by hydrolysis and decarboxylation (an acetoacetic ester synthesis), to assemble the carbon skeleton of 49. This choice mandates the inclusion of a carboxylic ester activating group in 58.

Woodward examined three different strategies to discover which route actually provides the best overall yields of 58 from 57. Each pathway exploits readily available starting materials. Interestingly, the longer (less convergent) route, using methyl acetate and methyl chloroacetate as building blocks, gave better overall yields than the other two routes that exploit symmetrical diester precursors. It is also significant that, in each route, a dissonant starting material is used to provide the dissonant circuit in 58.

Conversion of 58 to the key tricyclic intermediate 61 was then achieved by hydrolysis, decarboxylation, hydrogenolysis, chlorination, Friedel-Crafts acylation, Claisen condensation-Dieckmann cyclization, and then another hydrolysis and decarboxylation. The interesting selective demethylation that produced 60 results from intramolecular transesterification of an intermediate benzyl alcohol followed by hydrogenolysis of the resulting benzylic ester. Methylation of 60 was performed solely to facilitate purification.

Woodward anticipated different properties for the three carbonyl groups in 61. Thus, the β-dicarbonyl array (C-11 and 12) is "a stabilized vinylogous carboxylic acid system, while the third, like that in simple α-keto acids, should be both highly susceptible to addition reactions and readily enolizable. The latter property should confer high nucleophilic reactivity upon the adjacent methylene." This reactivity was exploited to append to 61 a precursor fragment for ring A, and then the dimethylamino group by polar reactions. The third carbonyl, having served its purpose, was then removed by reduction to α-hydroxy-ketone 63, activation by intramolecular transesterification, and reductive cleavage of the resulting lactone.

A fully functionalized acyclic precursor was then elaborated for ring A by acylation of a methyl malonamate carbanion with the mixed anyhdride 64. Dieckman cyclization, exploiting the nucleophilic reactivity conferred to the adjacent methylene by the carbonyl at position 12, produced ring A. The required stereochemistry at position 4a in 64 is produced during addition of dimethylamine to 62, which favors the more stable equatorial epimer of 63. Installation of the last functional group, the hydroxyl at position 12a was then accomplished oxidatively to deliver 45.

A More Convergent Strategy for Tetracyclines.
In another strategy for the total synthesis of 6-desmethyl-6-desoxytetracycline (45), as in the previous synthesis by Woodward, the target is simplified by removing the C-12a hydroxyl. Then, except for the polar activation afforded by the C-4 dimethylamino substituent, the polar reactivity provided by the remaining functional groups is entirely consonant along any circuit as shown in 65. The polar disconnection 65 ⇒ 66 separates the molecule into two large fragments united by a simple methylene bridge. Further bond disconnections disect the A ring into two straight chain precursors 67 and 68, that can be reunited by polar reactions. The synthetic equivalents that Muxfeldt utilized for the synthons 67 and 68 were 69 and 70a, respectively.9
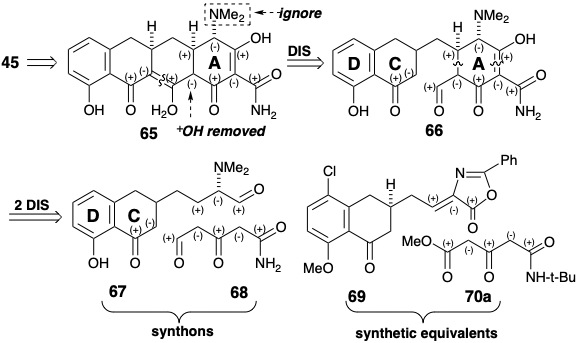
The highlight of the Muxfeldt synthesis is the ingenious reaction that produces the tetracyclic intermediate 71 in one step from the bicyclic precursor 69 in 82% yield! Final adjustment of functionality readily affords 45 from 71. The strategy benefits from a high degree of convergence. Moreover, 69 is readily available by an azalactone synthesis from 72 and 73. Finally, the intermediate CD-ring aldehyde 72 was prepared in good overall yield from 4-chloro-3-methylanisole.

The first total synthesis of a natural tetracycline, the 5-hydroxy derivative terramycin, was achieved by Muxfeldt.10 The A and B rings were assembled by the same strategy used above to generate 45. Thus, a CD-ring aldehyde 74, analogous to 72, was condensed with a preformed azathio lactone 75 analogous to an azalactone intermediate involved in the reaction of 73 with 72. The resulting Michael acceptor 76, analogous to 69 was then condensed with the unprotected amide 70b corresponding to 70a to generate the tetracycline ring system in one synthetic step. Subsequent deprotection of the masked hydroxyls and oxidative introduction of the last hydroxyl at position 12a was followed by removal of the thiobenzoyl masking group under exceptionally mild conditions upon treatment with methyl iodide. This reaction involves the generation and subsequent hydrolysis of a methyl thioimidic ester intermediate. Finally, controlled dimethylation of the primary amine gave terramycin.

The strategy employed for generating the key intermediate 74 exploits a temporary bridge in 78 to mask the aldehyde in latent form as an alkene and to create a folded tricyclic precursor that is expected to add a methyl nucleophile to the carbonyl at position 6 from the least sterically congested convex face. This assures a cis relationship between the methyl group and the neighboring bridgehead proton. The vinylogous α-diketone array in 77 is expected to be especially electrophilic, sufficiently activated that competition from addition to the acetate carbonyl can be avoided. Selectivity favoring addition to the carbonyl at position 6 rather than 11 may result from decreased electrophilicity owing to conjugation of the 11- but not the 6-carbonyl with the oxygen at postion 10. The presence of a cyclohexene in 77 recommends a cycloaddition synthesis involving a doubly activated electron-deficient quinone dienophile and relatively electron rich 1-acetoxy-1,3-butadiene.

In fact, the Diels-Alder reaction of acetoxybutadiene with juglone acetate (78) gave the diacetate 79 regioselectively, and addition of a methyl Grignard reagent to 79 is regio- and stereoselective. The cyclohexene ring, having served its purpose, was then oxidatively cleaved to generate an aldehyde from its latent equivalent, the carbon-carbon double bond. An unneeded two-carbon fragment was then removed by a sequence involving aldol condensation, oxidative cleavage, and finally retro Claisen cleavage of an intermediate β-diketone array in 80.

Because of a stereoelectronic preference for an endo transition state in the Diels-Alder reaction of 78, 79 is generated with the correct configuration at position 5. However, the oxidative cleavage that generates 80 produces a mixture of epimers at position 5. During replacement of the acetyl protecting group with a methoxymethyl, piperidine is used as a nucleophile to cleave the acetate and as a masking group to hide the sensitive aldehyde during alkylation of the phenol. Fortunately, hydrolysis of the enamine 82, by treatment with moist silica gel, generated epimerically pure 74 with the requisite configuration at position 5.