
4.7: Biosynthesis and Total Synthesis of Steroids
- Page ID
- 285643

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Biosynthesis of Lanosterol
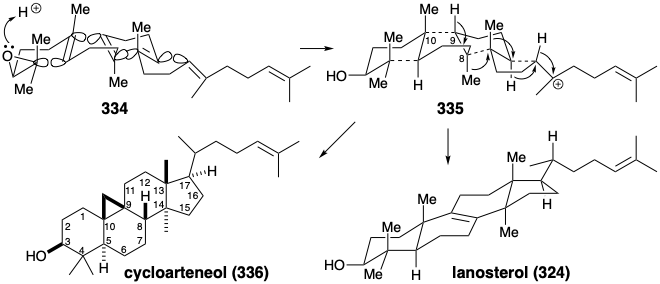
Lanosterol (324) is a member of steroid family of natural products. The isoprenoid origin of this triterpene, biogenetically a C30 hexamer of isopentenyl pyrophosphate, is not entirely evident from its structure. Thus, while two isoprene units are discernable in the right hand portion of 324 and two in the leftand portion, the ten carbon atoms in the central region of the molecule do not show isoprenoid connectivity. If, however, a methyl group were appended at position 8 or 9 of the four carbon 7,8,9,11-chain an isoprene unit would be formed. Since generation of the multicyclic carbon networks of terpenes occurs by electrophile-induced polyene cyclizations, the 8,9-C=C bond in 324 might arise by elimination of a proton from a carbocation precursor. A carbocation at the 8 position could have been generated by 1,2-migration of a methyl group from position 8 to a carbocation at position 14. Two more isoprene units are discernable in the central portion of the putative precursor 325. Topological analysis of 325 reveals six common atoms. Two disconnections between common atoms and two between a common and a noncommon atom greatly simplifies the structure suggesting an acyclic triterpene precursor 326 that might arrise by the head to head union of a diterpene pyrophosphate 327 with a monoterpene pyrophosphate 328. The strategy inferred above is close to that involved in the biosynthesis of lanosterol.
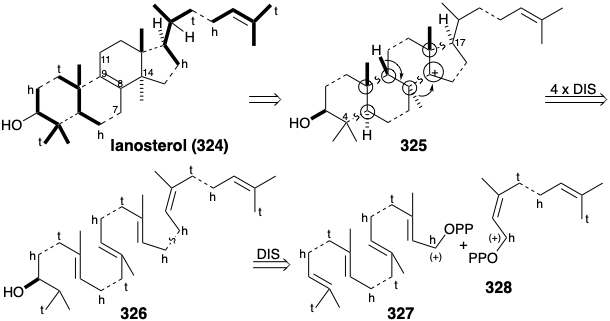
The actual biosynthetic strategy does indeed involve a head to head union of terpenoid pyrophosphates. However, a more efficient construction is achieved through a symmetrical acyclic triterpene intermediate, squalene (333), formed by the union of two molecules of a sesquiterpene precursor, E,E-farnesyl pyrophosphate (8). Since both carbons to be joined are electrophilic, polar formation of the central C-C bond of squalene cannot be direct. 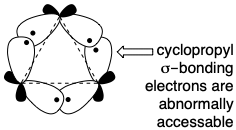 Rather, only the electrophilic activation provided by the functional group of one molecule of farnesyl-OPP is utilized to form a C-C bond with the nucleophilic C=C bond of a second molecule of farnesyl-OPP. Proton loss from the putative intermediate 3° carbocation 329 (or, possibly, the corresponding adduct with a nucleophilic moiety of the enzyme that catalyzes the process) produces a cyclopropylcarbinyl intermediate, presqualene pyrophosphate (330), that can be isolated from biosyntheses conducted in vitro in the absence of NADPH. In the presence of NADPH, 330 undergoes a reductive rearrangement formally involving a rearranged cyclopropyl 3° carbinyl cationic intermediate 331 and an allylic cation 332 that is captured by hydride to deliver squalene (333). The σ-bond in the cyclopropyl- carbinyl pyrophosphate 330 serves as nucleophile that displaces a pyrophosphate nucleofuge. The σ-bond electrons in cyclopropanes are especially accessible.
Rather, only the electrophilic activation provided by the functional group of one molecule of farnesyl-OPP is utilized to form a C-C bond with the nucleophilic C=C bond of a second molecule of farnesyl-OPP. Proton loss from the putative intermediate 3° carbocation 329 (or, possibly, the corresponding adduct with a nucleophilic moiety of the enzyme that catalyzes the process) produces a cyclopropylcarbinyl intermediate, presqualene pyrophosphate (330), that can be isolated from biosyntheses conducted in vitro in the absence of NADPH. In the presence of NADPH, 330 undergoes a reductive rearrangement formally involving a rearranged cyclopropyl 3° carbinyl cationic intermediate 331 and an allylic cation 332 that is captured by hydride to deliver squalene (333). The σ-bond in the cyclopropyl- carbinyl pyrophosphate 330 serves as nucleophile that displaces a pyrophosphate nucleofuge. The σ-bond electrons in cyclopropanes are especially accessible.
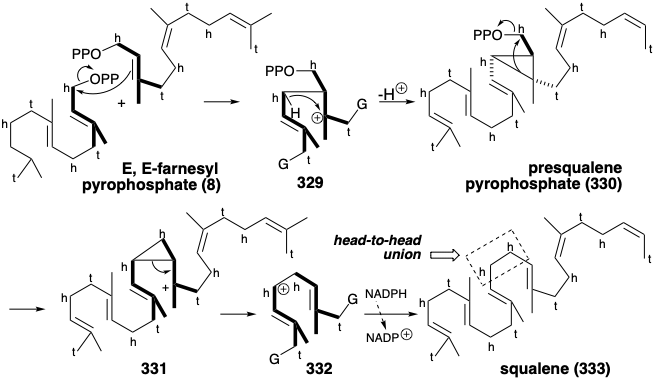
Asymmetric epoxidation converts the prochiral acyclic triterpene precursor 333 into a homochiral epoxide, squalene oxide (334). Generation of the four fused rings of lanosterol is then initiated by intramolecular alkylation of a C=C bond by a tertiary electrophile provided by protonation of the epoxide. A total of four consecutive alkylations, known as a polyene cyclization, deliver the putative intermediate 335 that rearranges by a series of two 1,2-hydride and two 1,2-methyl shifts to give lanosterol (324) after proton loss from the 9 position.
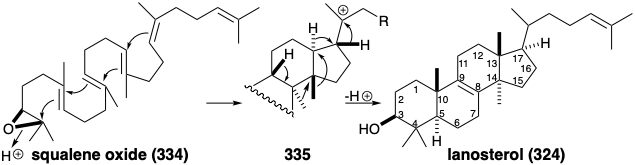
Especially interesting are the stereochemical details of the polyene cyclization and subsequent rearrangement of 335 to deliver 324. The polyene cyclization involves stereospecific anti-periplanar addition across three C=C bonds in 334 (see below). The folded conformation of 334, required for cyclization to 335, is probably imposed by the enzyme involved since appreciable steric congestion is present in both 334 and the intermediate 335. Relief of steric strain provides a large driving force for the rearrangement of 335 to 324 that involves stereospecific inversion of configuration at each stereocenter during 1,2-hydrogen or methyl shifts. 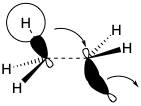 Thus, each 1,2-hydride or 1,2-methyl shift occurs to the backside of the orbital connected to the departing nucleofuge in what can be viewed as a series of nucleophilic substitution reactions – where σ-bonding electron pairs serve as the nucleophiles. Instead of proton loss to give 324, the rearrangement of 334 in some higher plants and algae ends with proton migration from the 9 to the 8 position and proton loss from the methyl group at position 10 forming a cyclopropane ring in cycloarteneol (336). This mechanism for the generation of a cyclopropane is analogous to that for the production of presqualene pyrophosphate (330) from 329.
Thus, each 1,2-hydride or 1,2-methyl shift occurs to the backside of the orbital connected to the departing nucleofuge in what can be viewed as a series of nucleophilic substitution reactions – where σ-bonding electron pairs serve as the nucleophiles. Instead of proton loss to give 324, the rearrangement of 334 in some higher plants and algae ends with proton migration from the 9 to the 8 position and proton loss from the methyl group at position 10 forming a cyclopropane ring in cycloarteneol (336). This mechanism for the generation of a cyclopropane is analogous to that for the production of presqualene pyrophosphate (330) from 329.
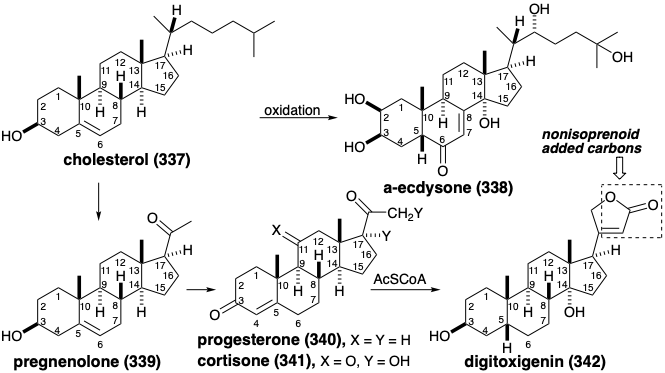
Nor and Seco Steroids. Although lanosterol (324) and cycloarteneol (336) are irregular triterpenes, i.e. their carbon skeletons are not composed of intact isoprene units, these triterpenes possesses the expected thirty carbons. Many steroids, that are derived biologically from lanosterol, contain fewer than thirty carbons and are referred to as nor triterpenes. Thus, for example, formation of cholesterol (337) from lanosterol (324) occurs by oxidative conversion of three methyl groups into formyl or carboxyl substituents that are lost as formate or cabon dioxide to give a tris nor triterpene after saturation of the side-chain C=C bond and migration of the Δ8,9 C=C bond to the Δ5,6 position. The biosynthesis of some steroids from cholesterol, such as the insect molting hormone α-ecdysone (338), simply involves oxidative introduction of functionality and C=C bond migrations. The loss of six carbons from the sidechain of cholesterol (337) leads to pregnenolone (339) the biosynthetic precursor of the female reproductive hormone progesterone (340) and the adrenocortical hormones such as cortisone (341) that is generated by oxidative functionalization of 340. The biosynthesis of the cardiac steroids such as digitoxigenin (342), that occurs in plants, creates the butenolide moiety by addition of a two carbon nucleophile from acetylCoA to the electrophilic side-chain carbonyl of 340.
Addition of a carbon atom as an electrophilic methyl group from S-adenosylmethionine (SAM) occurs during the biosynthesis of ergosterol (343). A pericyclic rearrangement of 343 to 344 followed by a thermally allowed antarafacial [1.7]-sigmatropic rearrangement of hydrogen from the methyl at position 10 to the 9 position leads to the generation of vitamin D2 (345). Sigmatropic shifts of hydrogen necessarily involve positive overlap, i.e., with the same phase, of the hydrogen half-occupied σ-orbital with the ends of the highest occupied pentadienyl π-orbital Both 344 and 345 are seco steroids, i.e. their carbon skeletons lack one of the ring C-C bonds of the tetracyclic steroid skeleton.

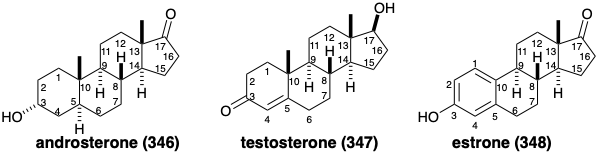
The entire cholesterol sidechain is removed during biosynthesis of the male sex hormones androsterone (346) and testosterone (347) and biosynthesis of the female reproductive hormone estrone (348) even requires loss of the angular methyl substituent from position 10. Noteworthy is the fact that the biosynthetic strategy for 348 is exceptionally circuitous and lengthy considering the structural simplicity (only four centers of chirality) of this target. The justification, of course, is the availability of the starting material, the ubiquitous biological steroid precursor, cholesterol. Furthermore, Nature has at its disposal a vast armementarium of selective reagents (enzymes) to achieve surgically clean removal of unwanted carbon atoms or groups by activation of C-H bonds, even those that are remote from functionality.
Total Syntheses of Estrone. All circuits between the oxygen functionality in the A and D rings of estrone are consonant except those involving carbons 15 and 16. Therefore, activating functionality is essential to allow polar C-C bond formation between the D ring carbonyl carbon and carbon 16. Disconnection of this bond in 349 suggests a diester 350 which still has functional dissonance, e.g. between the two carboxyl groups. All dissonance is removed by shortening the propionic to an acetic sidechain as in 351. Polar disconnection of an ester stabilized nucleophile sugggests a β-keto ester 352 and further polar disconnections then suggest 353 and a methyl electrophile, as precursors. Finally, polar disconnection of 353 suggests a monocyclic aromatic precursor 354 with an entirely consonant side chain in which the added carbonyl at the incipient 9 position can facilitate further polar disconnections to 355, an ester stabilized nucleophile, and a glutaryl electrophile. The foregoing retrosynthetic analysis provides a linear strategy for the synthesis of estrone that starts with an intact A ring and then builds the B, C, and D-rings in succession by polar reactions.
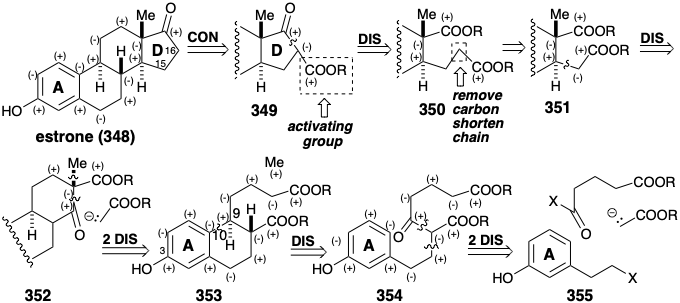
The first total synthesis of estrone, was completed21 in 1948. It assembled a methyl ether 357, related to 354, from the bromoethylanisole (356) by alkylation and then acylation of diethyl malonate. Cyclialkylation of the resulting ketotriester 357 makes use of the polar activation provided by target related functionality at position 3 that is conjugated through the aromatic ring with position 10. Hydrolysis and decarboxylative elimination then delivered alkene 35822 that upon hydrogenation, O-methylation, Dieckman cyclization, and C-methylation delivered an epimeric mixture from which the required ketoester 360 was isolated by fractional crystallization. Once again target related functionality, here the incipient carbonyl at position 17, facilitates polar bond formation. It is also noteworthy that target unrelated functionality, a carbonyl group on the carbon that will become position 9, serves as a lynchpin for connecting major segments in the 356 to 357 conversion and for generating the last connection for the B-ring in the 357 to 358 conversion. Reformatsky condensation followed by dehydration and hydrogenation provided diester 362 and an epimer from which it was separated by fractional crystallization. The lack of stereocontrol in this synthesis of 362 necessitated tedious isolations from isomer mixtures and resulted in a low overall yield.
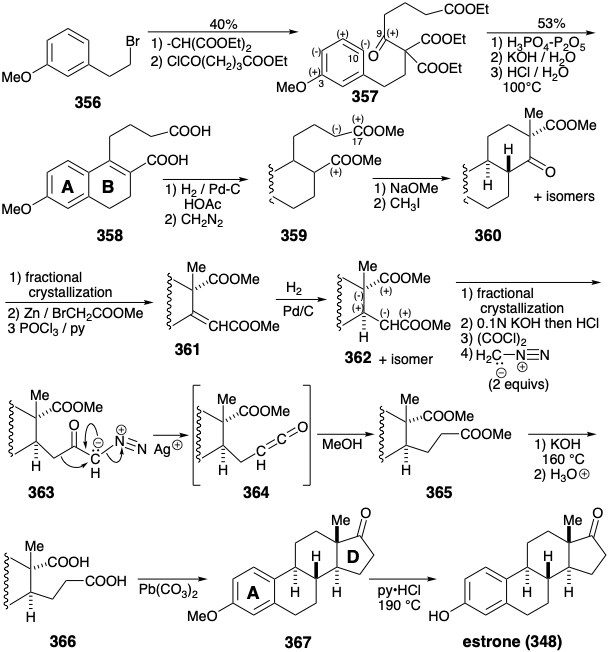
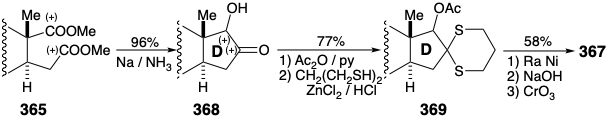
Chain elongation of the consonant 1,5-diester 362 generated a dissonant 1,6-diester precursor 365 of the dissonant D-ring cyclopentanone in 367. The creation of a dissonant product by polar reactions requires a dissonant reactant. In the present case this role is played by diazomethane that is dissonant by virtue of the presence of a biphilic activating group. Thus, the diazonium group stabilizes an α carbanion providing nucleophilicity for C-C bond formation with an acyl chloride and subsequently serves as a nucleofuge promoting migration of a nucleophilic alkyl group from the carbonyl carbon in 363 to the neighboring carbon in 364.
An alternative construction of the dissonant D-ring cyclopentanone in 367 from the consonant 1,5-diester 365 generates a C-C bond between the two electrophilic ester carbons by a nonpolar process, reductive coupling.23 Thus, an acyloin condensation provides 368 from which the unneeded carbonyl is removed by reductive desulfurization of the derived thioacetal 369.
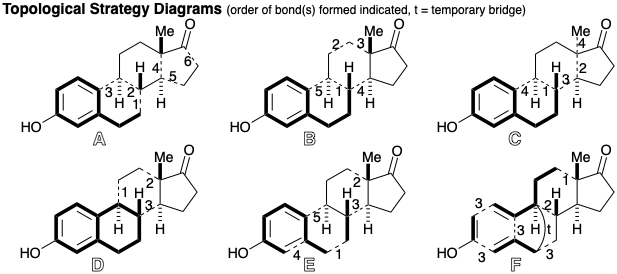
Topological Analysis of Estrone. The topological strategy for a synthesis can be summarized in a diagram that shows the starting material with bold outline and bonds formed in completing the skeleton with dashed lines. The two previous estrone syntheses illustrate strategies featuring late construction of the dissonant D-ring as in diagram A which allows the sequence of C-C connections employed. Greater efficiency can be accomplished by incorporation of the dissonant D-ring as a preformed unit as in B. Almost as efficient is the use of a dissonant precursor from which the D-ring is readily generated immediately after assembling an intermediate containing all the carbon atoms required for the skeleton as in C. A highly convergent and, therefore, exceptionally short synthesis is achievable by joining a preformed AB-ring unit with a D-ring unit as in D. The strategies summarized by A-D use aromatic starting materials for the aromatic moiety in the synthetic target. This tactic benefits from the relative stability of aromatic systems by avoiding potential yield-decreasing side reactions that might occur during manipulation of nonaromatic intermediates containing more reactive functionality. Interestingly, some efficient modern syntheses, summarized by E and F, generate the A-ring after assembly of a nonaromatic precursor containing all the skeletal carbons of the final target. Further discussion of the strategies B-F is deferred to a full consideration of each synthesis.
A strategy summarized topologically by B introduces nontarget related functionality, carbonyl groups in 370 on the carbons that will become positions 9 and 14, to activate the polar union of two pairs of common atoms and polar union of a symmetrical D-ring nucleophile with 371. Dislocation of this enone to a saturated ketone 372 with a nucleofuge (Nu) β to the carbonyl and the latter to an alkyne 373, reveals the possibility of exploiting a terminal alkyne nucleophile to assemble 373 from an arylpropyl electrophile 374.
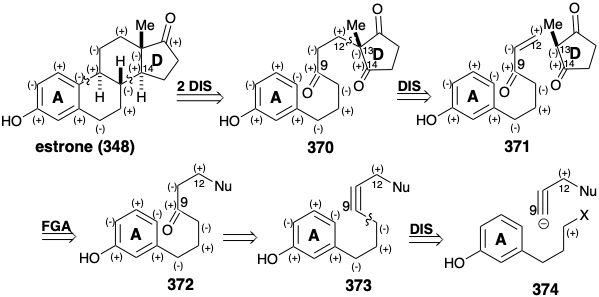
The use of a preformed D-ring in the dissonant building block 380 makes the synthesis24 more convergent. Greater efficiency is also provided by the consecutive formation of two connections, between the 8 and 14 and then between the 9 and 10 positions, in a single reaction that produces 382 from 381. Noteworthy is the regioselectivity of the 377 to 378 conversion. Clearly the diethylamino group provides a regiocontrolling influence, perhaps owing to a coordinative interaction with the \(\ce{Hg^2+}\) catalyst or to inductive destabilization of the development of a vinyl cation β to the electronegative nitrogen. Generation of the requisite stereochemical relationships is achieved by SAC during the delivery of hydrogen to 382 and TC during protonation of the preferred conformations of anionic intermediates in the reduction of 383.
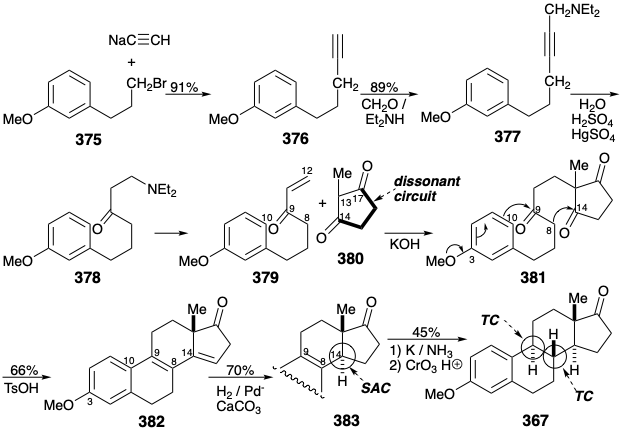
Another more recent synthesis,25 summarized topologically by C , is closely related to the B topological approach discussed above. Thus, the topology and polar reactivity involved in the 381 to 382 conversion is the same as that in the 387 to 388 conversion. However, a preformed D-ring is not exploited. Rather, this moiety is present in latent form in 386 that contains a dissonant circuit between masked 1,4-dicarbonyl groups. The dissonance derives from a latent 1,4-dicarbonyl array that protected by the aegis of aromaticity in the furan ring of 384 that is readily prepared by lithiation and then alkylation of α-methylfuran.

Stereoselective generation of the trans disubstituted C=C bond in 387 is accomplished by the Schlosser modification of the Wittig olefination. Thus, the β-oxidophosphonium intermediate 390, that is the major product from the addition of ylide 385 to an aldehyde, is converted to an epimeric β-oxidophosphonium intermediate 393 by protonation of a β-oxido ylide 392. This carbanion is thermodynamically favored over the epimeric carbanion 391. Subsequent syn elimination (perhaps through 2πs + 2πa cyclo- elimination of \(\ce{Ph3P=O}\) from a betaine intermediate) delivers the trans alkene stereospecifically.

Although 387 has no polar functionality at position 9, the 9-position participates as electrophile and the 8 position as nucleophile during polar cyclization to 388 in analogy with the 381 to 382 bis annelation. The stereoselectivity of this cyclization derives from a stereoelectronic preference for anti periplanar addition to the C=C bond in 387 and steric approach control during bond formation between positions 8 and 14. Stereoselective introduction of the angular methyl is accomplished by generation of the α-epoxide 389. Here SAC favors β attack by \(\ce{Cl^+}\) on the C=C bond. This is followed by stereospecific configurational inversion during intramolecular SN2 displacement. Finally, 1,2-migration of methyl during a pinacol rearrangement generates the required β-methyl configuration and a trans CD ring junction.
The most efficient strategy for total synthesis of estrone is also the most convergent, joining a preformed AB-ring unit 394 with a D-ring unit 380 as in D. This, the Velluz synthesis26, provides an industrial source of estrone that is more economical than biosynthesis. The polar union of 395 with 380 epitomizes the efficient use of functionality to facilitate skeletal construction. Interestingly, 380 is a vinylogous carboxylic acid sufficiently acidic to protonate 395. The resulting carbocation 396 is stabilized by target related functionality at position 3 while nucleophilicity at position 13 is stabilized by the target related oxygen functionality at position 17 in the enolate 397. Target related functionality at position 3 also facilitates generation of the 8-14 C-C bond during acid catalyzed cyclization of 400 to 382. A stereoselective route from 382 to estrone methyl ether (367) was described above.
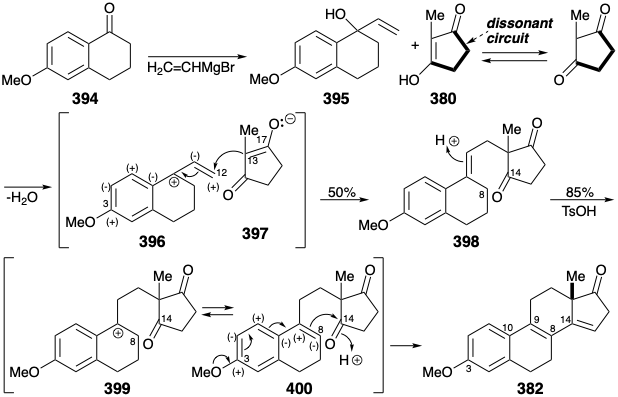
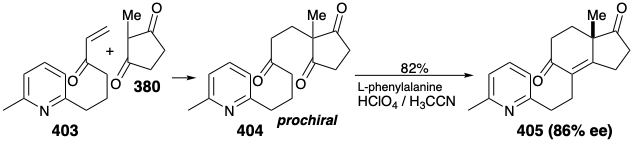
An Enantioselective Synthesis. The foregoing strategies all produce racemic estrone that must be resolved to provide the natural enantiomer. An enantioselective synthesis of natural (+)-estrone was reported in 1975.27 The key step converts prochiral trione 404 into enantiomerically enriched dione 405 by an enantioselective aldol condensation. Except for the use of a pyridine ring as a nonnucleophilic latent 1,5-dicarbonyl precursor of the estrone A ring, the topological and polar strategy is identical with that described above involving Michael condensation of 379 with 380. In the present synthesis, all the carbons required for the final target are united in a Michael condensation of 403 with 380. After the crucial enantioselective generation of ring C, the required trans CD ring junction is established by steric approach control during catalytic hydrogenation of an unsaturated alcohol derived from 405 by selective hydride reduction of the more electrophilic unconjugated carbonyl. Completion of the last two skeletal connections required unmasking of a latent 1,5-dicarbonyl array by Birch reduction of 406 followed by base catalysed hydrolysis of the resulting 1,4-dihydropyridine intermediate and aldol condensation. Hydrolysis of the ketal at position 9 then allows equilibration of the C8 stereocenter, and generation of the final skeletal connection by another aldol condensation. Aromatization of 408 delivers crude estrone. A single recrystallization gave optically pure (+)-estrone (13% from 380) as well as racemic estrone (3% from 380) from the mother liquor.
Estrone Synthesis by Cycloadditions. Since the B ring of estrone (348) contains one C=C bond in a six-membered ring, a thermal cycloaddition synthesis is possible. An intramolecuar Diels-Alder reaction could provide estrone from 409. Furthermore, another pericyclic rearrangemant can be employed for the synthesis of 409. Thus, the 1,3-diene array in 409 can be generated by electrocyclic rearrangement of a cyclobutene 410.
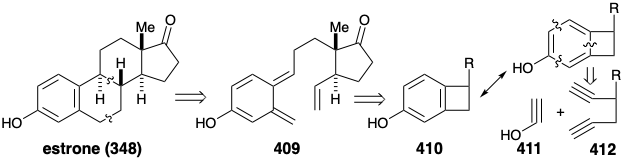
One particularly striking application28 of this strategy29 generates a benzocyclobutene intermediate 416 in an intramolecular cobalt(I)-catalyzed alkyne cyclotrimerization involving 1,5-diyne 412 with a synthetic equivalent 413 of the ketene enol 411. The trimerization is actually a stepwise process involving oxidative addition to cobalt(I) to generate a cobalt(III) cyclopentadiene 414. Diels Alder cycloaddition of 413 with 414 then produces 415 from which the cobalt(I) catalyst is regenerated by a reductive elimination that delivers 416.
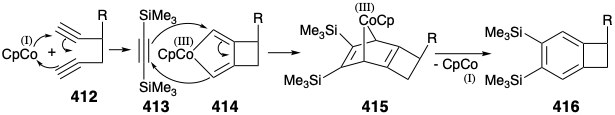
The correct relative configurations at positions 13 and 14 were established by steric approach control during alkylation of an enolate that was produced regiospecifically by 1,4-addition to 417 and regenerated from 418. Cycloaddition of 419 with bis(trimethylsilyl)acetylene in the presence of cyclopentadienylcobalt dicarbonyl generated an intermediate benzocyclobutene 420. Heat promoted generation of the derived ortho xylelene followed by intramolecular cycloaddition afforded 421 in 71% yield overall from 419. The stereoselective generation of the correct configurations at positions 8 and 9 arises from a preference for an exo transition state in the Diels-Alder cycloaddition. Steric congestion destabilizes the alternative endo transition state that would otherwise be favored by secondary orbital overlap. Mono protodesilylation of 421 removed the 2-silyl group with a 9:1 preference over the 3-silyl group. Oxidative desilylation generated the 3-hydroxyl and delivered estrone in 24% overall yield from 2-methyl-2-cyclopenten-1-one (417) in seven steps.
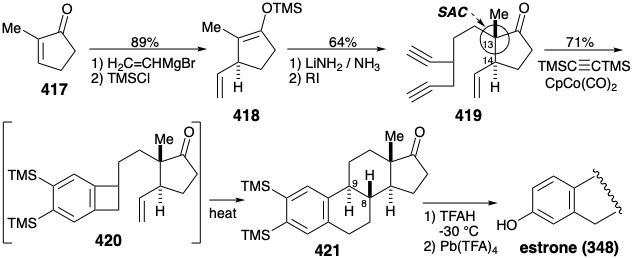
Total Synthesis of Cortisone. The strategy for the first total synthesis30 of cortisone (341) focused on the problem of assembling a trans fusion between the C and D rings. The challenge was to overcome the thermodynamic preference for a cis fusion between a five and a six-membered ring. Since a trans fusion is favored thermodynamically between two six-membered rings, an attractive solution seemed to be to create an intermediate with a six-membered D ring and to contract the six to a five-membered ring. With this goal in mind, an aldehyde group at position 17 in a cyclopentene precursor 422 ought to provide a starting point for building the sidechain found at this position in cortsone. Polar disconnection between carbons 16 and 17 of the D ring suggests a dialdehyde precursor 423 that would provide 422 by intramolecular aldol condensation. Connection of the two reactive aldehyde groups in 423 suggests a latent dialdehyde precursor 424 containing a six-membered trans-fused D ring.
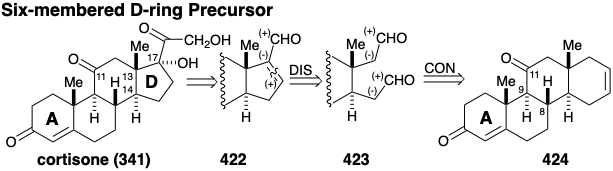
Disconnection of two bonds in the A ring at the ring fusion results in a major topological simplification. The ring fusion carbons in the A ring of 424 are common atoms, and disconnection of these two bonds, each between a common and a noncommon atom, completely removes the A ring. All circuits between the two carbonyl groups in 424 are dissonant. Therefore, both carbonyls cannot be used simultaneously to provide polar activation for generating C-C bonds. Rather, additional functionality, e.g. a carbonyl group at position 5, must be added to a precursor 425 to provide electrophilic activation at position 5 and nucleophilic activation at position 10 that can be exploited in conjunction with target related functionality at position 3 to assemble the A-ring. Such a polar cyclohexenone annelation process, the "Robinson annelation", had recently been devised in which an alkyl vinyl ketone serves first as an electrophile at the β-position of the vinyl group and then as a nucleophile at the α' position. Thus, two- step polar condensation of 425 with methyl vinyl ketone could be expected to provide the A ring in 424. A similar process could also be used to add the B ring to a bicyclic precursor 426. Because the carbonyl group at the 11 position in cortisone cannot provide the polar reactivity required for the annelation described above, its introduction can be delayed until the latter stages of the synthesis.
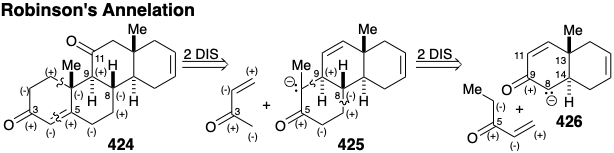
The cyclohexene unit in 427 suggests a cycloaddition synthesis involving a Diels-Alder reaction between a C ring precursor dienophile and 1,3-butadiene. However, generation of a trans ring fusion would require a C ring precursor containing a severely strained trans C=C bond in a six-membered ring. A more reasonable strategy would be to generate the thermodynamically favored trans ring fusion by epimerization of a cis fused precursor that would be produced, in turn, by a Diels-Alder reaction of a cis C=C bond in a dienophile. The carbonyl at the 9-position in 427 could be present in latent form in a precursor 428. A carbonyl group at position 8 in 428 could provide the polar activation required for epimerization of a cis ring fusion in 429 into a trans ring fusion. This carbonyl would also activate the conjugated C=C bond in 430 toward Diels-Alder reaction with a relatively electron rich diene. However, this strategy is fatally flawed because 430 can be expected to aromatize by enolization to afford 431. To block aromatization and provide additional activation of the dienophile, a second carbonyl can be added at position 12. This suggests a p-quinone dienophile 434 that would deliver a cis fused adduct 433 by Diels Alder reaction with 1,3-butadiene, and a trans fused intermediate 432 by epimerization.
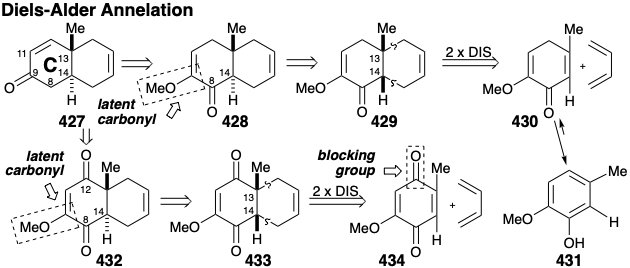
The more electron deficient methyl-substituted C=C bond in 434 is expected to be a more reactive dienophile towards 1,3-butadiene than the more electron rich methoxy-substituted C=C bond.
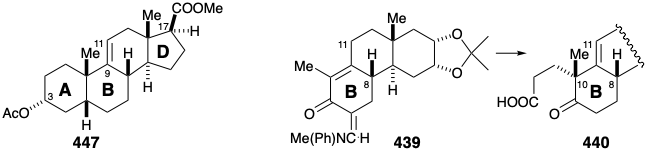
After having provided activation for epimerization at position 14 in 433, the carbonyl substituent at position 8 in 432 is removed by reduction to a hydroxyl, acetylation, and reductive cleavage. Simultaneously, the blocking carbonyl group at position 12 is removed by reduction and dehydration affording 427. Nucleophilic activation at position 8 is then enhanced by adding a formyl group that exists in the enol form in 435. Robinson annelation with ethyl vinyl ketone (EVK) then creates the B ring in 437. Thus, after facilitating the Michael alkylation, the activating formyl group is removed by a retro Claisen condensation upon treatment with \(\ce{KOH}\). Simultaneously, \(\ce{KOH}\) catalyzes an intramolecular aldol condensation in 436 generating the B ring in 437.
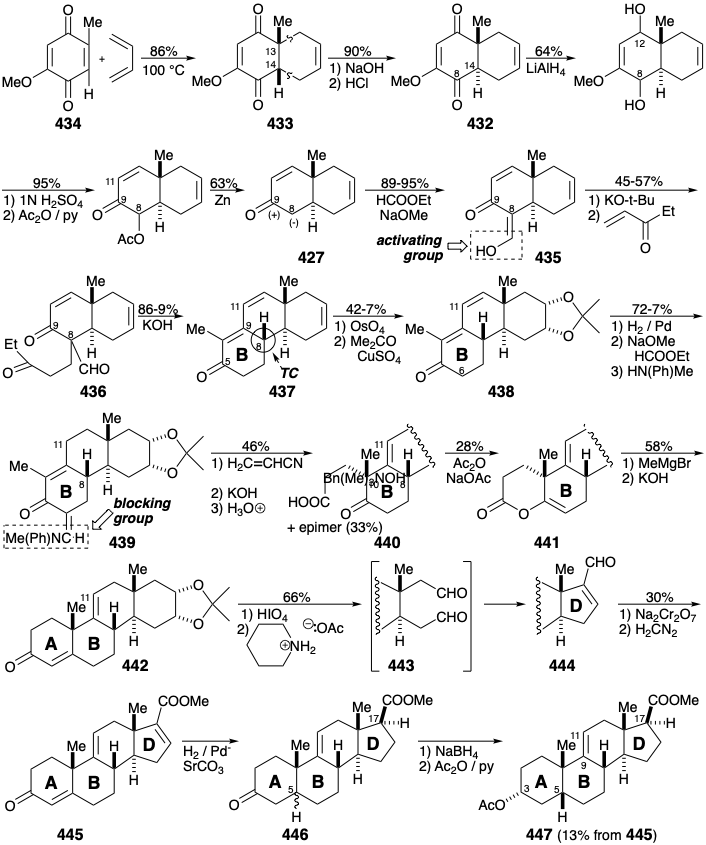
The stereoselectivity of this annelation can be ascribed to a thermodynamic preference for the requisite configuration at position 8 that is epimerizable owing to conjugation with the carbonyl group at position 5. Selective dioxidative addition to the unconjugated C=C bond in triene 437 is feasible because the electrophilic \(\ce{OsO4}\) is less reactive toward the electron deficient conjugated C=C bonds. Selective saturation of the sterically less encumbered, less substituted C=C bond in 438 is followed by installation of an enamine derivative of a formyl substituent as a blocking group at position 6 in 439. 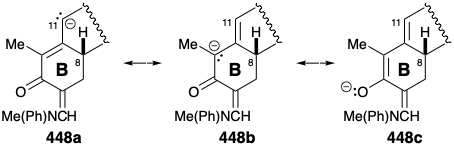 The contrasting applications of formyl substituents in 436 and 439 is noteworthy. Of the three remaining acidic hydrogens in 439, proton abstraction from position 11 is least sterically encumbered. The resulting ambident dienolate nucleophile, as expected, is selectively Michael alkylated α to the carbonyl group owing to greater electron density at the central compared with the terminal carbon atom of the 1-oxa-pentadienyl array in the intermediate carbanion. In other words, the resonance form 448b is more important than 448a of 448c.
The contrasting applications of formyl substituents in 436 and 439 is noteworthy. Of the three remaining acidic hydrogens in 439, proton abstraction from position 11 is least sterically encumbered. The resulting ambident dienolate nucleophile, as expected, is selectively Michael alkylated α to the carbonyl group owing to greater electron density at the central compared with the terminal carbon atom of the 1-oxa-pentadienyl array in the intermediate carbanion. In other words, the resonance form 448b is more important than 448a of 448c.
Unfortunately, alkylation of 448 is nonstereoselective producing carboxylic acid 440 and an epimer after hydrolysis of an intermediate nitrile. The final carbon required for the A ring of cortisone was to be provided by \(\ce{MeMgBr}\). However, to prevent addition to the ketone carbonyl in 440, the latter group was masked as an enol lactone in 441. The methyl ketone produced by addition of \(\ce{MeMgBr}\) to 441 delivered 442 by intramolecular aldol condensation. The poor yield for the 440 to 442 conversion would be improved, no doubt, by modern methods. Thus, a chemoselective reaction of the acid chloride from 440 with \(\ce{LiMe2Cu}\) would provide a high yield of the corresponding methyl ketone.
Contraction of the D-ring was accomplished by oxidative cleavage to dialdehyde 443 that gave 444 by a remarkably regioselective aldol condensation. Saturation of the two least sterically encumbered C=C bonds in the derived ester 445 gave a mixture of epimers 446 (at the 5 position) separation of which was best achieved after conversion to the 3-hydroxy derivative that then provided 447 by acetylation.
At this point the total synthesis intersected with "extensive prior investigations by many groups on the partial synthesis, from natural sources, of cortisone." Thus, introduction of the 11 carbonyl group was accomplished by epoxidation of alkene 447, hydrolysis of the epoxide to triol 449, and oxidation to dione hemiketal 450. Nucleophilic replacement of the oxygen at position 9 by Br proceeded with allylic rearrangement upon treatment of 450 with \(\ce{HBr}\). Selective generation of a trans A-B ring fusion and the required configuration at position 9 in 451 is a consequence of thermodynamic control. Reductive debromination of bromoketone 451 followed by reduction to a diol, selective acetylation of the less sterically encumbered hydroxyl at position 3, and oxidation delivered the 11-keto steroid 452.

Assembly of the dissonant cortisone side chain was accomplished by polar condensation of the acid chloride from 452 with a dissonant building block, diazomethane, and reaction of an intermediate diazoketone with acetic acid. Saponification of the resulting diacetate 453 and selective reacetylation of the primary hydroxyl delivered 454. Introduction of a hydroxyl group at position 17 was then accomplished by 1,2-dioxidative addition to 455. Steric approach control favored the requisite configuration at the 17 position. Finally, introduction of unsaturation between carbons 4 and 5 was accomplished by bromination followed by dehydrobromination of 456. Dehydrobromination of α-bromoketones can be complicated by α' proton abstratction that leads to a Favorskii reaction through the formation of a cyclopropanone. The mild Mattox-Kendall method avoids this pitfall because α-bromo hydrazones readily eliminate bromide to generate intermediate α,β-unsaturated hydrazones, e.g. 457. Pyruvic acid effects removal of the hydrazine while hydrolysis of the side chain acetate occurred upon treatment with \(\ce{HCl}\) completing the first total synthesis of cortisone (341) by R. B. Woodward in 1952.

As a practical source of supply for medicinal applications, Woodward's synthesis could not compete with partial syntheses from other natural steroids that are readily available from plants. A major shortcomming of the total synthesis is the generation of a racemic product. The natural enantiomer was available by resolution of the intermediate 445, but at least half of this precious advanced intermediate was discarded, i.e. the wrong enantiomer. Although the biosynthesis of cortisone is circuitous, it produces only one enantiomer owing to an entirely enantioselective epoxidation of squalene.
An Enantioselective Strategy for Cortisone. The achievement of an industrially viable total synthesis of cortisone depended on the development, in 1966, of an enantioselective strategy.31 Interestingly, the strategy evolved from methods and intermediates developed during a synthesis of cortisone that employs methoxytetralone (394) as a BC ring precursor32 in contrast to the Velluz estrone synthesis mentioned earlier (see section 5.3) that used this starting material for the AB ring moiety. But the key development in the evolution of an enantioselective strategy was the conception of a prochiral starting material and a route for its elaboration into a steroid.33 Thus, extensive research on the partial synthesis of cortisone from naturally derived 17-ketosteroids had established methods that allow the elaboration of the cortisone sidechain from precursors like 458.
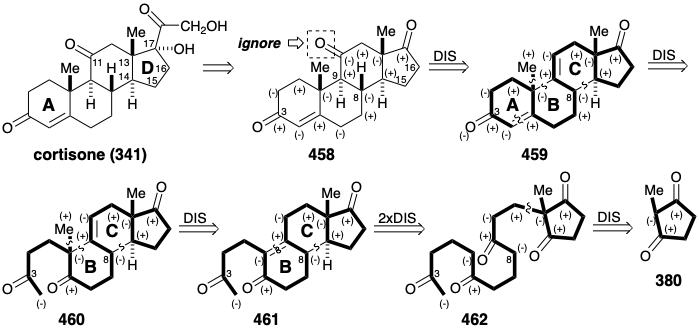
Polar analysis of this subtarget, ignoring the 11-keto group, reveals that all circuits between the oxygen functionality in the A and D rings are consonant except those involving carbons 15 and 16. Introduction of the 11-keto group by addition to a C ring C=C bond in a precursor 459 is precedented by the similar functionalization of 447 (see section 5.4) in the Woodward synthesis.
Polar disconnection of the 4,5-C=C bond in 459 suggests intramolecular aldol condensation of 460 for generating the A ring. In the Woodward synthesis, a similar intermediate, i.e., 440, was generated by alkylation of 439. Methylation of the analogue 461 of 439 might similarly provide 460. Further polar disconnections of the B and C rings suggests a monocyclic precursor 462 that is achiral. We saw a similar intermediate in Danishefsky's enantioselective synthesis of estrone. Thus, 404 (see above) contains two of the carbonyl groups of 462 in latent form. In the estrone synthesis, asymmetry was introduced by an enantioselective aldol condensation of 404 to give 405. Also, in that synthesis, 404 was prepared from an intact D-ring precursor 380 by Michael addition to enone 403.
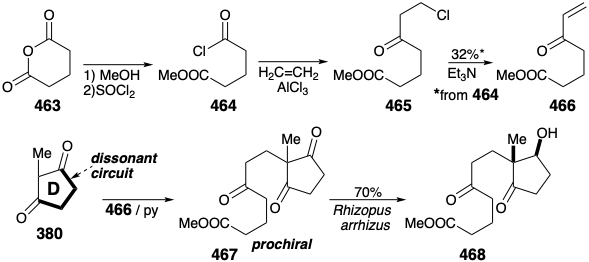
A similar strategy was employed a decade earlier to prepare an analogous prochiral precursor for cortisone. However, as we shall see, a different enantioselective transformation of the prochiral precursor 467 (see below) was used to introduce asymmetry into this synthesis of cortisone by Velluz.
A symmetrical starting material 463 is converted to the Michael acceptor 466 through Friedel-Crafts acylation of ethylene with the acid chloride 464 and dehydrochlorination of the intermediate β-chloro ketone 465. Although the yield of 466 is poor, it is readily available in kilogram quantities from inexpensive starting materials. Condensation of 466 with the intact D ring precursor 380 delivers 467. Enantioselective microbiological reduction provides the optically pure mono reduction product 468 in good yield as a single diastereomer.
Acid-catalyzed intramolecular aldol condensation then creates the C ring in 469. Stereoselective saturation of the C=C bond in 470 creates the requisite trans CD ring fusion, without a need for the lengthy ring contraction process of the Woodward construction of the trans CD ring fusion. To achieve selective polar connection at the carboxyl carbon in 470 of a nucleophilic fragment containing the remaining carbons required for the A and B rings, the ketone carbonyl must be masked. This is accomplished intramolecularly by enol esterification to give 471.
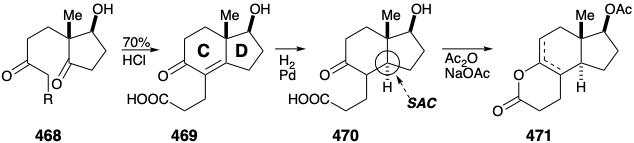
Grignard reagent 472 reacts with 471 to deliver 473 that is cyclized by intramolecular aldol condensation to provide the B ring in 474. The correct configuration at position 8 is assured by thermodynamic control because this center is conjugated with the carbonyl at position 5. Nevertheless, proton abstraction from 474 occurs primarily at the 11 position leading upon angular methylation to 475 completely stereoselectively with the requisite configuration for cortisone. The stereoselectivity of this alkylation contrasts sharply with the lack of stereoselectivity observed in the corresponding 384 to 385 conversion in the Woodward synthesis. The contrasting stereochemical behavior of these two alkylations are a consequence of mechanistic differences. Thus, the alkylation of 439 is a reversible, thermodynamically controlled, Michael reaction whereas the methylation of 474 is a kinetically controlled process. Axial methylation from the β face of the enolate is stereoelectronically preferred because it leads directly to the chair conformer of 475.
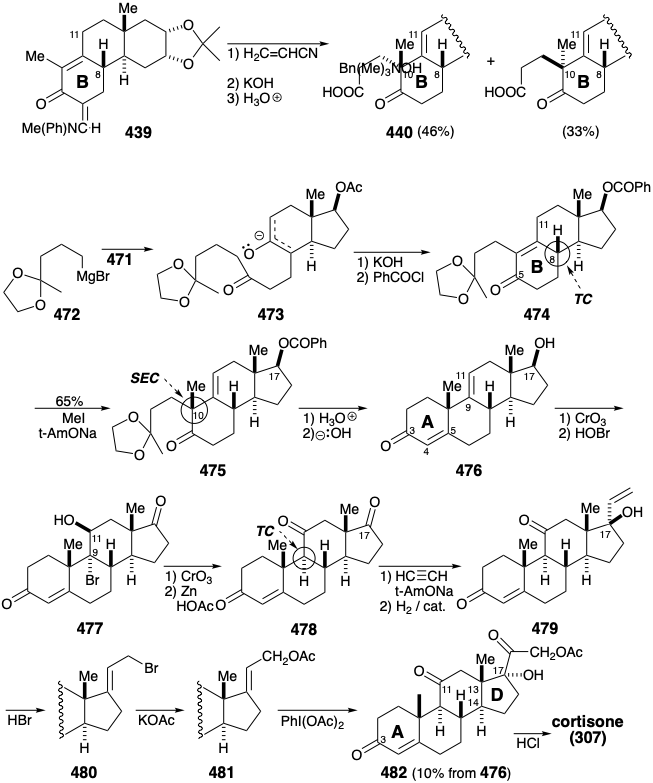
Generation of the A ring is then accomplished with a third intramolecular aldol condensation providing 476 after hydrolysis of the 17 benzoate. Oxidation of the 17 hydroxyl, and introduction of oxygen at the 11 position by addition of \(\ce{HOBr}\) provides 477 that, upon oxidation and reductive debromination, delivers 478 stereoselectively with the thermodynamically preferred configuration at position 9 which is α to the carbontyl at position 11. Finally, elaboration of the cortisone side chain was accomplished by ethynylation of the more electrophilic unconjugated 17 carbonyl. Catalytic hydrogenation then provided the tertiary allylic alcohol 4 7 9 . Bromodehydroxylation of 4 7 9 occurred with allylic rearrangement, presumably through an allylic cation intermediate, while replacement of \(\ce{Br}\) in 480 with \(\ce{OAc}\) proceded by a direct SN2 substitution. The unusual oxidizing agent, phenyliodosodiacetate and a catalytic quantity of \(\ce{OsO4}\), converted the more nucleophilic C=C bond in 481 directly into the α hydroxy ketone 482.