
4.6: Biosynthesis and Total Syntheses of Diterpenes - Spatol
- Page ID
- 285454

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Biosynthesis
As for all natural products, a successful synthetic strategy for spatol existed before any human endeavor. It is always interesting to examine Nature's strategy because an analogous approach, a biomimetic strategy (mimicing Nature), may be effective in the laboratory. Thus, the diterpenes are C20 compounds derived biogenetically from E,E,E-geranylgeranyl-PP (192) or its geometrical isomers.
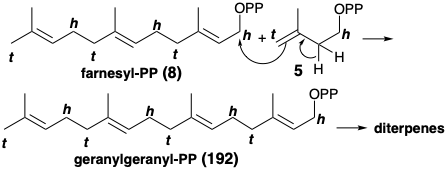
These acyclic tetramers of Δ3-isopentenyl-PP (5) arise from reaction of 5 with a trimer such as E,E-farnesyl-PP (8). Subsequent intramolecular electrophilic addition of the allylic pyrophosphate to the trisubstituted C=C bonds can lead to various mono and multicyclic carbocations such as 193. Another general route to carbocationic electrophiles involves protonation of C=C bonds. A hypothetical pathway for the biosynthe- sis of spatol (196) involves protonation of a C=C bond and intramolecualr electrophilic addition of the resulting carbocation to produce a cyclobutane (195) by proton loss from 194. This hypothesis derives support from the natural occurrence of 195.14 The oxygen functionality in 196 is presumed to arise from oxidative metabolism of 195 by the marine organisms that produce this tricyclic diene and a wide variety of oxygenated metabolites with the spatane carbon skeleton.

Topological Analysis of Spatol
Ex post facto topological retrosynthetic analysis of the biosynthetic strategy reveals an important feature. The tricyclo[5.3.0.02,6]decane nucleus of the spatane diterpenes incorporates 4 common atoms (circled in 196), the four atoms of the B-ring. The biosynthetic strategy benefits from the powerful topological simplification that accrues from removing bonds between two sets of common atoms, 4-8 and 9-10. This suggests a monocyclic topological synthon 197. For this synthon, one synthetic equivalent, 198, is suggested by our biogenetic hypothesis.

Another synthetic equivalent, 199, is suggested by the possibility of an intramolecular 2π + 2π cycloaddition. An expeditious synthesis15a of the sesquiterpene b-bourbonene ( 2 0 1 ) exploits intramolecular photocycloaddition of germacrene D (200) an intermediate analogous to 199. UV evidence (λmax = 259 mm, ε = 4500 in n-hexane) indicates a significant transannular interaction between the two endocyclic C=C bonds in the sesquiterpene 200. Thus, 200 probably prefers a conformation in which the two endocyclic C=C bonds are situated parallel and face to face with each other, and the isopropyl substituent occupies the less sterically hindered equatorial configuration. Thermodynamic control of the conformation of 200 assures the proper configuration at the isopropyl bearing carbon while stereoelectronic control (syn periplanar = suprafacial addition) assures the correct cis,anti,cis configurations at the cyclobutane stereocenters. An analogous photocyclization to generate a precursor 202 for spatol is not favorable since a substituent R which is to become the side chain must occupy a more sterically encumbered axial position as in 202a rather than the more thermodynamicaly favorable conformation 202e.

A Topological and Stereochemical Strategy
A different topological disection of the cis-anti-cis-tricyclo[5.3.0.02,6]decane nucleus of β-bourbonene (201) has also been exploited for its synthesis. Thus, removing two bonds between pairs of common atoms can generate two cyclopentene precursors, 203 and 204, that could be united by a 2πs + 2πs photocycloaddition (see section 3.3). In fact, UV irradiation of 2-cyclopenten-1-one with 204 results in a photocycloaddition that is orientationally nonselective, producing a 1:1 mixture of structural isomers 205 and 206.15b However, the cycloaddition is favorably stereoselective owing to a steric approach controlled preference for cycloadddition to the face of the cyclopentene ring opposite the isopropyl substituent. This stereoselectivity detracts from the utility of a similar synthesis for spatol because the allylic diepoxide side chain in spatol (196) is cis to the cyclobutane rather than trans as is the isopropyl group in 201 or 205.

Thus, the allylic diepoxide side chain or its precursor in a cyclopentene intermediate 207 can be expected to favor the wrong stereoselectivity in a photocycloaddition with 2-cyclopenten-1-one, i.e., favoring 208 or 209 rather than the desired adduct 210 or its structural isomer 211.

One strategy to surmount this shortcoming of cyclopentene photocycloadditions for the total synthesis of spatol uses a temporary bridge to shield one face of a cyclopentene ring to preclude addition to that face.16 Thus, such a bridge can be provided by linking the hydroxyl group at the 5-position with a carboxymethyl group that also serves as a progenitor of the sidechain at position 7 as in lactone 213. Furthermore, the lactone can be derived from a latent precursor, ketone 214, by a Baeyer-Villiger oxidation. Double disconnection of 214 by a cycloelimination suggests photocycloaddition of a norbornenone 215 with cyclopent-2-en-1-one. Thus, in 214 a temporary oxoethano bridge shields the α-face of the incipient C-ring enforcing stereoselective cycloaddition of the A-ring precursor cyclopent-2-en-1-one trans to the incipient 5-hydroxyl group.

This strategy has one obvious flaw. The strained bicyclic homoallylic ketone 215 was expected to readily undergo photoinduced cleavage to diradical 216. However, masking of the carbonyl in 215 would circumvent this problem and facilitate differentiation between the two carbonyl groups in the photocycloadduct 214. Also, the configuration at the 7-position in 212 would have to be inverted to provide the requisite configuration at this stereocenter in spatol. A synthesis of 6-methylbicyclo[2.2.1]hept-5-ene-2-one (215) from vinyl acetate and methyl-1,5-cyclopentadiene is possible through a Diels-Alder reaction of these starting materials. Although the reaction produces a mixture of structural isomers, saponification followed by oxidation gives a mixture of isomeric ketones from which 215 can be isolated by distillation.

Masking a Sensitive Ketone
Masking of the carbonyl group in 215 proved unexpectedly difficult. Only a moderate yield of the ethylene ketal 217 was available by acid-catalyzed ketalization of 215 under conditions which give an excellent yield of ketal from the 6-unsubstituted analogue, bicyclo-[2.2.1]hept-5-en-2-one, owing to a competing fragmentation to 218. The proclivity of 215 toward this fragmentation undoubtedly arises from the relative stability of the tertiary carbocation 220 and the relief of ring strain attending conversion of 219 to 220.

An unusual choice for the masking group was developed during a search for a group that could be introduced under nonacidic conditions. Thus, ketone 215 is converted quantitatively into an epimeric mixture of cyanohydrin silyl ethers 221 by reaction with trimethylsilyl cyanide. While the use of an unsymmetrical masking group might seem unwise since this leads to epimeric mixtures of several intermediates, this is a small price to pay for the otherwise ideal characteristics of the cyanohydrin silyl ether masking group. Thus, photocycloaddition with cyclopent-2-en-1-one delivered two epimeric adducts 222x and 222n with high stereo (cis,anti,cis ring fusions and exo addition to the bicyclohept[2.1.1]ene) and orientational (cyclopentane carbonyl remote from the bridgehead methyl group) selectivity. Serendipitously, the major adduct 222x crystallized from the photoreaction mixture together with the dimer of cyclopentenone from which it was readily separated by trituration with hot hexane leaving behind pure dimer.  Pure 222x was then obtained in 51% yield, based on 2 2 1 , by elution of the partially purified product through a column of silica gel with ethyl acetate- hexane. Column chromatography of the hexane soluble photoproduct afforded a fraction from which nearly pure minor cycloadduct 222n crystallized together with a little 222x. This mixture is suitable for Wittig olefination to produce methylidene ketone 224, vide infra. The combined isolated yield of 222x plus 222n exceeds 60%. A sample of pure 222n was obtained by HPLC. The epimeric relationship between 222x and 222n was demonstrated by production of the same diketone 223 upon hydrolysis of the cyanohydrin silyl ether masking group and also by production of the same methylidene ketone 224 upon reaction with methylenetriphenylphosphorane followed by hydrolysis of the cyanohydrin silyl ether. The conversion of 222 into 224 was performed as a one-pot procedure affording pure 224 in 89% overall yield. The utility of the cyanohydrin silyl ether masking group in the above transformations is noteworthy. It is introduced under mild neutral reaction conditions. It is sufficiently robust to survive UV irradiation, chromatography on silica gel, and Wittig olefination; but it is readily converted to a carbonyl group by the aqueous base generated upon addition of water to the Wittig reaction mixture.
Pure 222x was then obtained in 51% yield, based on 2 2 1 , by elution of the partially purified product through a column of silica gel with ethyl acetate- hexane. Column chromatography of the hexane soluble photoproduct afforded a fraction from which nearly pure minor cycloadduct 222n crystallized together with a little 222x. This mixture is suitable for Wittig olefination to produce methylidene ketone 224, vide infra. The combined isolated yield of 222x plus 222n exceeds 60%. A sample of pure 222n was obtained by HPLC. The epimeric relationship between 222x and 222n was demonstrated by production of the same diketone 223 upon hydrolysis of the cyanohydrin silyl ether masking group and also by production of the same methylidene ketone 224 upon reaction with methylenetriphenylphosphorane followed by hydrolysis of the cyanohydrin silyl ether. The conversion of 222 into 224 was performed as a one-pot procedure affording pure 224 in 89% overall yield. The utility of the cyanohydrin silyl ether masking group in the above transformations is noteworthy. It is introduced under mild neutral reaction conditions. It is sufficiently robust to survive UV irradiation, chromatography on silica gel, and Wittig olefination; but it is readily converted to a carbonyl group by the aqueous base generated upon addition of water to the Wittig reaction mixture.
Amplifying SAC
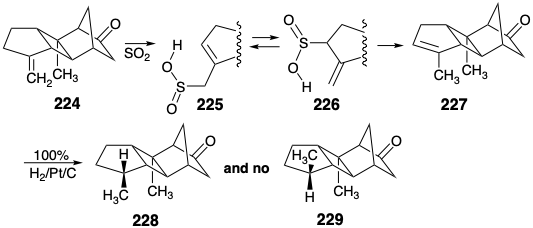
Catalytic hydrogenation of 224 favored the required epimer 228 over the useless byproduct 229 by 10:1 owing to steric approach control by the methyl group in 224 that shields the α-face of the A-ring. However, separation of 228 from the mixture could not be achieved by any method except fractional crystallization, and this only allowed isolation of the desired epimer in only fair yield. To circumvent this separation problem, 224 was isomerized to the endocyclic alkene 227 by \(\ce{SO2}\). This clean, quantitative isomerization presumably involves ene addition of \(\ce{SO2}\) to 224 producing 225. Subsequent [1.3] sigmatropic rearrangement of sulfur affords 226 that undergoes retro ene fragmentation delivering 227. Catalytic hydrogenation of 227 delivers 228 cleanly and quantitatively. Apparently the closer proximity of the endocyclic C=C bond to the methyl group in 227 than the exocyclic C=C to the methyl group in 224 results in greater steric hindrance to α hydrogen delivery in 227 than in 224.
Resolution
Efficient, virtually quantitative resolution of ketone 227 was readily achieved by flash chromatography and crystallization of the 1,2-adduct with chiral lithiosulfoximine 230. Retro ene elimination of the less soluble dextrorotatory diastereomer (+)-232 delivered ketone (+)-227 that was correlated with (+)-spatol by conversion to a degradation product from natural spatol (vide infra). The sulfoximine (+)-(S)-231 was recovered in 96% yield.

A Temporary Bridge During Hydride Delivery
Catalytic hydrogenation of (+)-227 provided ketone (+)-228. Introduction of oxygen at position 5, cleavage of the temporary bridge, and inversion of configuration at the 7-position were then addressed. Thus, Baeyer-Villiger oxidation to give lactone (+)-233, saponification, and methylation of the resulting acid provided alcohol (+)-234. Masking of the 5-hydroxyl provided (-)-235 that was carboxylated to give malonic ester (-)-236. Epimerization at the 7-position was initiated by selenenylation followed by oxidative deselenenylation of the resulting 237 to deliver alkylidene malonic ester 238. Reduction of 238 with \(\ce{NaBH4}\) followed by removal of the MEM protecting group with \(\ce{TiCl4}\) afforded a 2:1 mixture respectively of the cis hydroxy malonic ester 240 and its C-7 epimer, the desired trans hydroxy malonic ester 241. This disappointing result suggested that the 2-methoxyethoxymethoxy (OMEM) substituent at the 5-position sterically hinders hydride delivery to the α- face of the C=C bond in 238.

A remote hydroxyl group was found to foster pseudointramolecular syn hydride delivery via an alkoxyborohydride intermediate 242. Thus, treatment of the derived hydroxy alkylidene malonate (+)-239 with \(\ce{NaBH4}\) delivered the desired trans hydroxy malonic ester (-)-241 completely stereoselectively. The overall yield was 9% from C-ring precursor 215 in 21 steps including the resolution.

Enantiospecific Synthesis with a Chiral Auxillary
A different synthesis18 of a homochiral tricyclodecylmalonic ester intermediate 245 (an analogue of 241) was designed with a focus on exploiting butenolide 243 as a chiral auxiliary to establish the correct absolute configuration during generation of the B-ring by a 2π + 2π photocycloaddition with A-ring precursor 244. Homochiral butenolide 243 is readily available from L-glutamic acid. An allylic oxygen substituent in 244 provides a point of attachment for the malonic ester side chain and activation for the introducing oxygen at the 5-position.

In a retrosynthetic format, the strategy envisions attachment of the malonic ester last by a stereospecific SN2 alkylation with 246. Since the chiral auxiliary 243 does not provide the cyclopentane ring required for the A-ring of 245, this ring will have to be generated after the photocycloaddition of 250.

The A-ring might be created by an intramolecular aldol condensation of a bis methylketone precursor 248. This step is potentially flawed because an undesireable alternative aldol condensation is possible. However, an excellent precedent is provided by a similar step in a total synthesis of the sesquiterpene α- bourbonene (F). Thus, intramolecular aldol condensation of A delivers cyclopentenone E and none of the isomeric cyclopentenone C. Apparently, cyclization to the aldol condensation product B is disfavored by steric hinderance by the angular methyl substituent in the alternative aldol condensation product D. Steric approach control (SAC) should favor β-delivery of hydrogen during reduction of 247 to give the requisite configuration at position 1 in 246. The proper orientation during generation of the B-ring can be assured by a temporary bridge, an ester, between 243 and the C-ring precursor 244.

Intramolecular photocycloaddition of the ester 250 from the chiral auxiliary 243 delivered cyclobutane 251. Addition of a methyl group, the one carbon needed to complete the A-ring required 4-10 steps depending on the configuration of the C-7 substituent in 251. Reduction of the remaining ester in 252 then provided the methyl group at position 4 in 253 which had been functionalized solely to allow construction of the temporary bridge in 250. Functional group manipulation then provided dione 254 which underwent completely selective aldol condensation affording 255. Stereoselective hydrogenation of 255 created the stereocenter at position 1 and removed the benzyl protecting group. Wolff-Kishner reduction of the resulting saturated ketone delivered 256. Introduction of the 5-hydroxyl and 7-malonic ester substituents then required oxidation to 257, reduction and Mitsunobu inversion to give 258, stereoselective epoxidation followed by hydride reduction, protection, and deprotection to deliver 259 and nucleophilic substitution which provided malonic ester 245 in 0.6% yield overall from 243 in 29-35 steps.

Convergent and Linear Strategies
The overall yield of homochiral malonic ester (-)-241 from racemic (±)-215 in the first synthesis was 9%, more than an order of magnitude higher than the 0.6% overall yield of homochiral malonic ester 245 from homochiral 243 in the second synthesis. The success of the first synthesis, that relies upon resolution to introduce asymmetry, is especially noteworthy because resolution is inherently inefficient -- it provides at best as 50% yield of the correct enantiomer. Two factors diminish the penalty for using resolution. First, the resolution is performed very early in the first synthesis and, therefore, the effort wasted by discarding half of the racemic product is minimized. Second, Johnson’s sulfoximine method is spectacularly effective. Furthermore, the advantages of the clever plan to exploit the readily available chiral auxiliary 243 to introduce asymmetry into the second synthesis cannot overcome the penalty arising from the absence of a methyl group at position 4 or a hydroxyl group at position 5, and the lack of an A-ring in the photocycloadduct 251 from 243. The first synthesis is more convergent than the second. Thus, two large fragments are constructed that contain most or all of the skeletal atoms and functionality of the target and these fragments are then united. Such an approach has several advantages over a linear synthesis, that is one in which the molecule is constructed by sequentially uniting many small fragments or introducing functionality after skeletal construction is complete. A convergent synthesis is more efficient as measured by overall yield. If the average yield of an n-step synthesis is Ψ%, then the overall yield will be 100(Ψ/100)n%. A 21-step linear synthesis with an average 95% yield will have an overall 34% yield, or an overall 11% with an average 90% yield, or an overall 0.9% with an average 80% yield. In contrast for a convergent synthesis that combines two intermediates each prepared by 10-step syntheses (i.e. a total of 21 steps), the overall yield will be 56% with an average 95% yield, or an overall 31% with an average 90% yield, or an overall 9% with an average 80% yield. In effect the convergent synthesis is only 11 steps. The two abovementioned syntheses are a case in point. The average yield per step, 84-87%, in the second synthesis was almost as high as the 89% average per step yield in the first synthesis. The 15 fold lower overall yield for the second synthesis is almost entirely the consequence of its greater length, 29-35 steps versus 21 steps.
Stereocontrolled Sidechain Construction
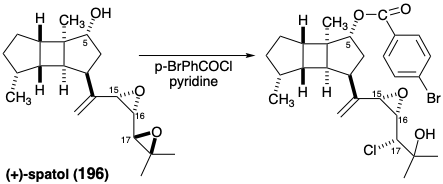
Two strategic challenges must be met for completion of a total synthesis of spatol (196). First, the unique allylic vicinal diepoxide in 196 was presumed to be highly electrophilic because epoxide ring opening by chloride, a weak nucleophile, accompanies esterification upon treatment of 196 with p-bromobenzoyl chloride and pyridine. Second, the three stereocenters of the flexible sidechain must be assembled with the correct configurations relative to those in the rigid tricyclic nucleus.
The malonic ester group in the intermediates (-)-241 and 245 could provide a three-carbon allylic precursor of the spatol side chain. Koga converted 245 into allylic alcohol 260 by a modified Marshall reduction.

However, attempted one-step conversion of (-)-241 into the allylic alcohol 263 by the Marshall reduction, i.e. LAH reduction of the sodium enolate, failed completely. Therefore, this transformation was accomplished by monosaponification to 261 and decarboxylative aldol condensation with formaldehyde to provide acrylic ester (+)-262. Hydride reduction then delivered allylic alcohol (+)-263 that was selectively oxidized with \(\ce{MnO2}\) to the aldehyde (+)-264. To correlate this synthetic intermediate with the natural product, (+)-264 was acetylated. The totally synthetic acetate showed [α]D22 +25.1° that compares well with the naturally derived acetate which showed [α]D22 +26.5°.1

An Absolute Asymmetric Strategy
Disconnection of both a nucleophilic oxygen and an electrophilic carbon from carbon 15 of spatol suggests a precursor 266 in which the sulfonium functional group provides the requisite biphilic reactivity at carbon 15.19 The correct relative configurations for the stereocenters in the tricyclic nucleus and at position 17 are assured in an absolute asymmetric synthesis by using building blocks 266 and 267 with the correct absolute configurations. Although very short, this convergent strategy provides no control over the configurations at positions 15 and 16.

Ylide 266 was prepared from allyic alcohol 260 through sulfonium salt 268. Reaction of ylide 266 with aldehyde 267 produced spatol in only 3% yield together with the isomeric allylic cis diepoxide 270 (1.5%) and a mixture of trans diepoxides 271 (8%). Moreover, 266 exists in equilibrium with an alternative ylide 269 that underwent [2.3] sigmatropic rearrangement producing the homoallylic sulfide 272 (35%) as the major product of the reaction.

A Stereospecific Epoxydiol Rearrangement Strategy
The last step in the reaction of ylide 266 with aldehyde 264 involves vicinal alkylation of an alkoxide during cyclization of 273. The reaction of aldehyde 277 with ylide 276 is a related strategy. The epoxy aldehyde 277 might be available from aldehyde (+)-264 by Corey-Fuchs alkynylation to give 278, homologation with formaldehyde, Lindlar reduction of the resulting propargyl alcohol, asymmetric epoxidation of the derived allylic alcohol, and Swern oxidation of the resulting epoxy alcohol. Alternatively, 278 might be homologated to 275. Then, after assymmetric reduction of this propargyl ketone, Lindlar reduction, and VO(acac)2-catalyzed epoxidation, heterocyclization of the resulting 274 might deliver (+)-spatol. However, these strategies are too long, and ring closures of intermediates such as 274 may be derailed considering the potential, inter alia, for E-1 elimination and transepoxidation. Thus, intramolecular attack of the alkoxide in 280 at the 2° 16-position to give 281 rather that at the 3° 18-position to give spatol might even be favored. Such transepoxidation reactions (Payne rearrangements) are well known. However, the rearrangement to 281 is reversible while heterocyclization of 280 would be irreversible. Nevertheless, E1 elimination to give 279 seemed a reasonable concern.

An alternative strategy is possible involving an epoxy alkoxide similar to 281 but with the positions of the nucleofuge and alkoxide exchanged. Thus, Payne rearrangement should produce 283 but the trans stereochemistry of the epoxide in 282 should virtually preclude direct attack of the alkoxide at the 15-position to produce a tetrahydrofuran. The allylic electrophile at position 15 in 283 should be particularly effective in alkylating the neighboring alkoxide producing an allylic diepoxide 284.

Furthermore, an efficient strategy for assembling an epoxydiol precursor 285 for 282 seemed feasible. Thus, 285 should be available by regioselective epoxidation of 286 which should, in turn, could be prepared by the union of a C15 electrophile (+)-264 with a C5 nucleophile 287.

In model studies, a method was sought to produce the allylic diepoxide array of the spatol side chain from appropriately activated derivatives of epoxydiol 285. Initial results were disappointing. Thus, activation of erythro-288 as a mesylate, erythro-289, followed by treatment with solid \(\ce{K2CO3}\) in boiling isopropanol delivered threo-290 by intermolecular SN2 displacement rather than the desired vicinal diepoxide by Payne rearrangement followed by heterocyclization. Since the tertiary hydroxyl group in erythro-289 appeared not to be sufficiently nucleophilic to displace the epoxy leaving group, conditions were sought that would generate an alkoxide from the tertiary hydroxyl. Treatment with t-BuOK in t-BuOH promoted a clean, stereospecific rearrangement and heterocyclization to deliver the diepoxide trans,erythro-292. A route from the erythro-288 to an activated derivative of the threo epoxy alcohol requires activation with concomitant inversion of configuration. This was accomplished by the Still modification of the Mitsunobu reaction. Thus, reatment of erythro-288 with \(\ce{Zn(OTs)2}\), diethyl azodicarboxylate, and triphenylphosphine, gave tosylate threo-291 that, upon treatment with t-BuOK in DMF, afforded the allylic diepoxide cis,erythro-292 in 82% yield.

The substitution reaction of mesylate erythro-289 with isopropanol to give threo-290 suggested that a similar substitution with tetrabutylammonium hydroxide might provide a route to the inverted alcohol. Instead, however, a high yield of diepoxide trans,erythro-292 was obtained. The unexpected stability of this allylic diepoxide toward hydroxide is especially interesting in view of the epoxide-cleaving substitution reaction of spatol with the less nucleophilic chloride anion that gives a chlorohydrin (vide supra). Apparently, the latter reaction is an acid-catalyzed epoxide opening induced by pyridinium hydrochloride, a byproduct of the acylation with p-bromobenzoyl chloride in the presence of pyridine.
In a first attempt to implement the plan, addition of vinyllithium 287 to aldehyde (+)-264 provided a 1:6 mixture of triols 295 and 296 respectively after desilylation of the intermediate monosilyl ethers 293 and 294. To control the regioselectivity of epoxidation, the triol 296 was selectively disilylated. Vanadium-catalyzed epoxidation of 297 was then directed to the 15,16-C=C bond by the remaining allylic hydroxyl. Since the major epoxide product was the erythro derivative 298e, selective activation of the less hindered 15-hydroxyl in the corresponding triol 298e was performed with inversion of configuration. However, treatment with base produced an allylic diepoxide 300 with a cis,anti,cis- tricyclo[5.3,0,02,5]decane nucleus. Thus, Wagner-Meerwein rearrangement of the cis,anti,cis-tricyclo[5,3,0,02,6]decane nucleus of 298e to give 300, apparently owing to an unintended activation of the 5-hydroxyl that accompanied the desired activation of the hydroxyl at the 15-position.

These results suggested that derivatives of epoxy triols 299e and 299t in which the hydroxyl at position 5 is masked were needed for generation of the spatol side chain without accompanying rearrangement of the tricyclic nucleus. The lability of the allylic diepoxide array in spatol (196) under acidic conditions limited the choice of derivatives to those with masking groups that would be removable under neutral or basic reaction conditions. The further requirement for stability towards a vinyllithium reactant and the presence of unsaturation in the synthetic target recommended p-methoxybenzyl (MBn) ether derivatives. The MBn masking group is removable under mild conditions by oxidative cleavage with DDQ. Therefore, the MBn derivatives erythro-308b and threo-308b of 298e and 298t were prepared from diol (+)-263.

The assignment of an S absolute configuration at the 15-position to the major epimer (-)-305b was based on correlation with natural (+)-spatol (vide infra). Epoxidation of the derived silyl ether (-)-307 provided a 2:7 mixture of threo and erythro epoxides 308b. The major isomer, erythro-308b, was converted into a cis, erythro diepoxide (-)-310 by conversion to a threo tosylate with inverted configuration at the 15-position followed by base-induced Payne rearrangement, heterocyclization, and finally by deprotection of the 5-hydroxyl. That (-)-310 was not spatol (196) was evident from its optical activity, [α]D = -10.0° in contrast with [α]D = +45.6° reported for the natural product. Small chemical shift differences, e.g. vinyl 1H NMR resonances at δ5.14 and 5.09, confirmed that (-)-310 is epimeric at positions 15, 16, and 17 with (+)-spatol which exhibits vinyl resonances at δ5.13 and 5.02.

The minor isomer, threo-308b, was converted into (+)-spatol (196) by monomesylation followed by base-induced Payne rearrangement, heterocyclization, and deprotection of the 5-hydroxyl. Each resonance in the 1H NMR spectrum of synthetic (+)-spatol coincided within 0.01 ppm with a spectrum of an authentic sample of natural spatol.

An Absolute Asymmetric Stereoconvergent Strategy
Both of the aforementioned strategies for the spatol allylic diepoxide suffer from inadequate stereocontrol. Thus, while the correct absolute configuration at position 17 in 273 is assured by using the correct enantiomer of 267, generation of the stereocenters at positions 15 and 16 in 273 is not selective. Similarly, although either epimer at position 15 in 308 can provide an activated derivative with the correct configuration, i. e. by activation with retention or inversion of configuration, generation of the stereocenters at positions 16 and 17 in 308 is not favorably selective.

Since either epimer of epoxydiol 312 can be converted stereospecifically into spatol, the synthesis is stereoconvergent, and stereocontrol at the 15-position is unnecessary for an efficient total synthesis.. The correct stereochemistry at positions 16 and 17 could be assured by an absolute asymmetric strategy that combines the homochiral C15 aldehyde (+)-304 and a homochiral C5 α-epoxy nucleophile 313.
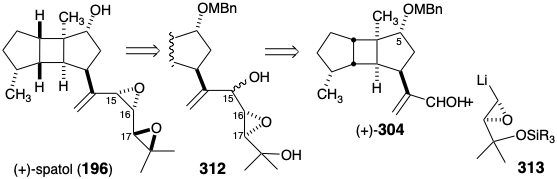
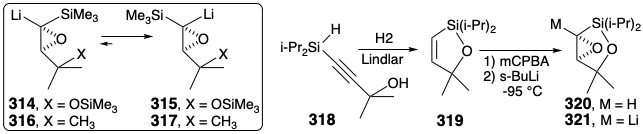
Because α-silyl epoxides are readily hydrodesilylated by moist fluoride with complete retention of configuration, a possible synthetic equivalent of synthon 313 is the silyl-stabilized α-lithioepoxide 314. However, a serious flaw could sabotage this strategy. Thus, although α-lithioepoxides are generally configurationally stable, the α-lithioepoxide 316, a close analogue of 314, exhibits an unusual configurational instability rearranging completely to 317 owing, no doubt to steric strain that is relieved upon trans-cis isomerization. A similar isomerization of 314 to 315 would derail the synthesis. This pitfall was circumvented by a temporary bridge between the C-silyl substituent and neighboring oxygen in 321. Thus, intramolecular O-silylation precludes isomerization of the carbanion 321 obtained by metallation of 320. Racemic 321 was prepared through epoxidation of a vinylsilane 319 that was generated by a novel hydrogenation-dehydrogenative-heterocyclization of 318.
In a model study, reaction of α-lithioepoxide 321 with 2-(i-propyl)acrolein delivered an epimeric mixture of adducts 322 favoring the erythro diastereomer by 7:3. Desilylation of erythro-322 gave epoxydiol erythro-323. Thus, the racemic intermediate 321 provides a two-step synthesis of epoxydiol precursors of the spatol allylic diepoxide. However, only conjunction of aldehyde (+)-304 with the correct enantiomer of 321 will provide the correct absolute configurations at positions 16 and 17 in 312 that are required to accomplish an efficient construction of natural spatol (196). A route to optically pure epoxide 321 must yet be found.
