
3.7: Levuglandins
- Page ID
- 285633

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
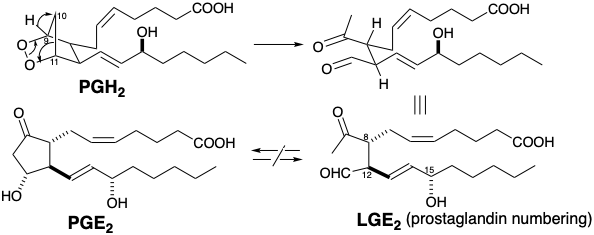
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Water-induced rearrangement of PGH2 occurs rapidly under the conditions of its biosynthesis to generate PGE2, PGD2, and two seco-prosta-glandin levulinaldehyde derivatives known as levuglandins (see section 4.1). Thus, for example, intramolecular hydride migration from the 9 to the 10-position in PGH2 accompanied by cleavage of the 10,11-C-C bond and the peroxide O-O bond generates levuglandin (LG) E2. This levuglandin is formally related to PGE2 by aldol condensation, although interconversion of PGE2 with LGE2 has never been observed.
Enantiocontrol with a Chiral Auxiliary
The availability of abundant supplies for biological testing and confirmation of the structure of LGE2 depended on the development of an efficient asymmetric total synthesis. Another strategy for deriving asymmetry from chiral nonracemic natural starting materials is to use it as a chiral auxilliary (vide infra), as illustrated by a synthesis of LGE2 from L-arabinose.23 A dominant consideration in planning a total synthesis of LGE2 is its proclivity toward dehydration, epimerization at positions 8 and 12 (prostaglandin numbering) and allylic rearrangement of the 14-15 (prostaglandin numbering) C=C bond into conjugation with the aldehyde carbonyl. Many of these difficulties are circumvented by replacing the aldehyde carbonyl with a latent equivalent, a vicinal diol as in 303. Conversion of 303 into LGE2 should be achievable under exceptionally mild conditions by oxidative cleavage with periodate. Stereocontrol is a more difficult challenge in the total synthesis of LGE2 than it was for the total synthesis of PGE2 since LGE2 is acyclic and, therefore, more conformationally mobile than PGE2 that has three of its four stereocenters arranged in the thermodynamically preferred all-trans configuration around a relatively rigid cyclopentanone ring.
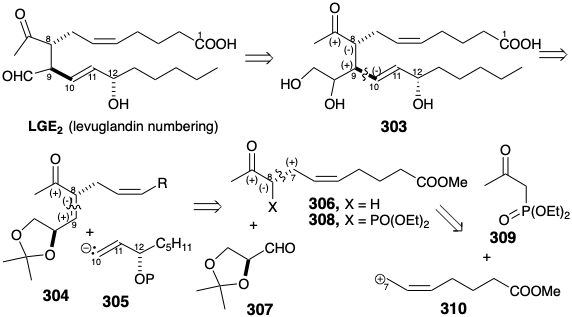
The tactic of using a vicinal diol as a latent aldehyde group apparently complicates rather than simplifies the synthetic target by adding a fourth stereocenter. On the contrary this additional center of chirality, that will not be incorporated in the final product, is the key to enantioselective generation of the correct absolute configuration at position 9 (levuglandin numbering). Furthermore, the correct configuration at position 8 should be available by epimerization of any 8-epi 303 that might be generated. Polar analysis of 303 suggests the possibility of exploiting electrophilicity at position 9 provided by the acetyl carbonyl as in 304 to allow polar bond formation with a chiral nonracemic nucleophilic vinyl synthon 305. Most importantly, such conjugate additions are highly diastereoselective. The neighboring alkoxy substituent is expected to foster generation of only the requisite absolute confuguration at position 9 during 1,4-addition of a vinyl cuprate nucleophile to 304. Further polar analysis of 304 suggests generation of this enone by aldol condensation of an enolate nucleophile with the aldehyde 307, L-glyceraldehyde acetonide. This chiral nonracemic starting material is readily available from L-arabinose (vide infra). Because its chiral center provides enantiocontrol but is not incorporated into the final product, 307 said to serve as a chiral auxiliary (a chiral unit that is incorporated into an intermediate to bias the stereoselectivity of one or more subsequent reactions after which it is cleaved from the substrate or its chiral center is removed). To activate enolate generation and control the regiochemistry of the aldol condensation, a diethylphosphono group is added to 306 as in 308. Further exploitation of the polar activation afforded by the acetyl carbonyl and phosphono groups should allow construction of 308 by allylation of 309 with the upper sidechain electrophile 310.
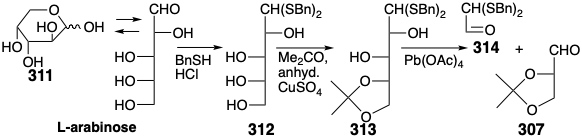
A synthesis of the chiral auxillary 307 from L-arabinose starts with interception of the acyclic aldose from its equilibrium with a pyranose form 311 by thioacetalization with benzylmercaptan. Selective ketalization of the resulting tetraol 312 delivers monoacetonide 313. Oxidative cleavage of the latter then produces 307 in admixture with 314 from which it is readily separated by distillation.24
A short, highly stereocontrolled, asymmetric total synthesis of LGE2 was executed23 from the commercially available 1-(diethylphosphono)-2-propan-one (309) that was allylated in good yield with bromoester 315. The main side reaction was diallylation. Horner-Emmons olefination of L-glyceraldehyde acetonide (307) with the carbanion derived from 308 delivers in excellent yield a mixture of geometric isomers E-304 and Z-304 in a 2.3:1 ratio. It is unnecessary to separate this mixture because either isomer reacts stereoselectively (i. e. SEC) with cuprate 316 to deliver an identical 7:3 mixture of 317 and its 8-epi diastereomer in excellent yield. This key reaction proved refractory. Little or no 1,4-addition could be achieved until it was discovered that anhydrous \(\ce{MgBr2}\) catalyzes the required reaction presumably by serving as a Lewis acid that enhances the electrophilicity of enone 304. Again separation of isomeric products is unnecessary because saponification of either diastereomeric ester 317 or 8-epi-317 generates an identical 7:3 mixture of the corresponding carboxylic acids. This is apparently the equilibrium ratio.
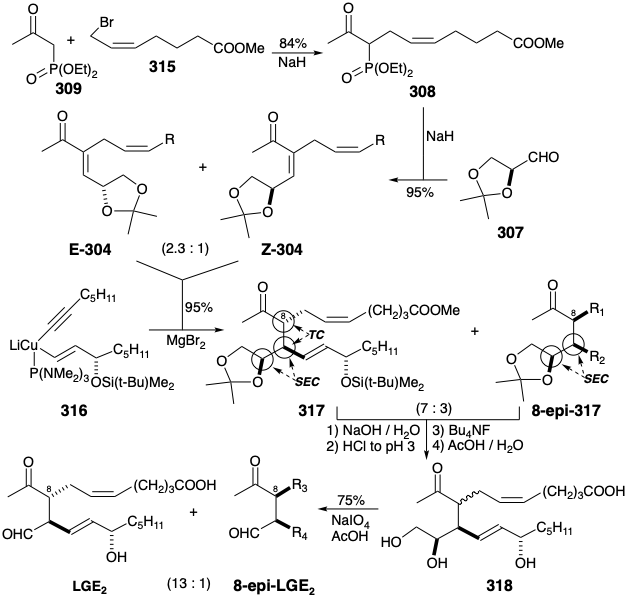
Most fortunately, separation of the diastereomeric acids was also unnecessary because either isomerically pure acid gave the same 13:1 mixture of LGE2 and its 8-epi diastereomer upon desilylation followed by acid-catalyzed hydrolysis of the acetonide and finally periodate cleavage of the resulting vicinal diol. The favorable diastereoselectivity of the acid-catalyzed epimerization that accompanied the deketalization of the vicinal diol was entirely unexpected. The vicinal diol also plays an important role in this serendipitous process. Thus, epimerization occurs in 318 but not in LGE2 or 8-epi-LGE2 under these conditions for hydrolysis and oxidative cleavage.