
3.6: Enantioselective Syntheses of Prostaglandins
- Page ID
- 285447

All of the foregoing total syntheses of prostaglandins produced these natural products together with their unnatural enantiomeric isomers because each synthesis began with nonasymmetric or racemic starting materials and employed racemic or nonasymmetric reagents. In some instances racemic mixtures of enantiomeric precursors were separated by resolution and then converted to the enantiomerically pure natural product. But this approach to the total synthesis of chiral nonracemic natural products is usually inherently wasteful since half of the racemic precursor, the wrong enantiomer, must be discarded. Very rarely, the wrong enantiomer can be converted to the correct enantiomer or converted into the natural product in an enantio-convergent synthesis by a unique reaction sequence.
There are three tactics that allow enantioselective synthesis of natural products. They all depend on the chirality of natural products to provide asymmetric starting materials or to induce asymmetry during the generation of chiral intermediates from prochiral precursors. Presented in the ensuing discussion will be examples of syntheses that are enantiocontrolled by the use of: (1) chiral nonracemic reagents; (2) microbial metabolism -- a special case of category 1; or (3) chiral nonracemic starting materials.
Enantiocontrol by a Chiral Nonracemic Reagent
In the synthesis discussed above, the chirality of 235 is determined by an intermediate 246 that was generated by reaction of an allyl nucleophile with the prochiral epoxide 240. Since the allylating reagent employed was nonasymmetric, the chiral product was racemic. The intermediate 235 has also been prepared in another manner, one that generates only the correct enantiomer required for the total synthesis of natural optically pure prostaglandins.19 In this asymmetric synthesis, the chirality of 235 is determined by an intermediate 247 produced by hydroboration of the prochiral diene 248 with a chiral nonracemic dialkyl borane.
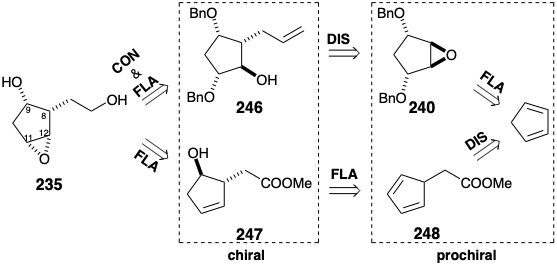
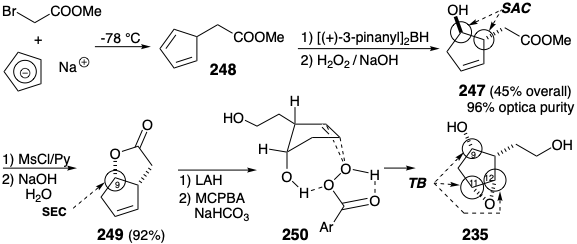
The 5-substituted 1,3-cyclopentadiene 248 was generated at -78° C by alkylation of sodium cyclopentadienide and treated in situ with (+)-di-3-pinanylborane, followed by alkaline hydrogen peroxide to yield hydroxy ester 247, that was at least 96% optically pure. Owing to steric approach control during the syn addition of boron hydride to 248 and subsequent stereospecific retention of configuration during oxidative replacement of boron with oxygen, the new stereocenter at position 9 (PG numbering) in 247 is generated stereoselectively with the unnatural configuration, opposite to that required for prostaglandins. However, the required cis configuration is readily generated by SN2 inversion of the hydroxyl, and lactonization delivers 249. The stereoselective generation of the cis epoxide in 235 depends upon the influence of a temporary bridge. Thus, the hydroxyl at C-9 directs the delivery of oxygen by hydrogen bonding with MCPBA as shown in 250. This temporary bridging in the transition state of a reaction is an example of a neighboring group effect.
Enantiocontrol by Microbial Metabolism
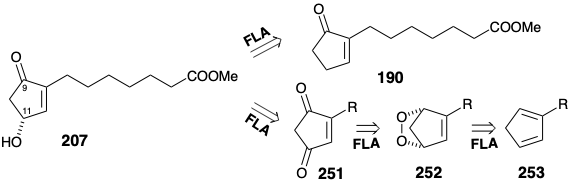
The enzymes which catalyze microbial oxidations and reductions are chiral nonracemic molecules that often promote highly enantioselective transformations of synthetic prochiral substrates.20 Several strategies have been explored for enantioselective microbial generation of hydroxycyclopentenone intermediates for syntheses of prostaglandins. For example, the chiral intermediate 207 might be available by microbial allylic oxidation of the prochiral precursor 190 or by microbial reduction of the prochiral precursor 251. The dione 251 should be readily available by oxidation of the hydroxycyclopentenone mixture obtained by base catalyzed disproportionation of a singlet oxygen 2π + 4π cycloadduct 252 with cyclopentadiene 253.
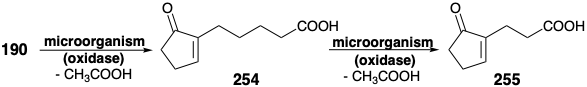
A flaw in the allylic oxidation strategy resulted from a general proclivity of fungi containing hydroxylases to degrade the carboxylic side chain of 190 by the β-oxidation-retro Claisen cleavage mechanism discussed in section 3.1. Thus, cleavage of one acetate unit produced 254 while 255 was generated upon loss of a second acetate.
A proclivity of dione 251 toward monoreduction is expected owing to the activating effect of opposed electron withdrawal by two conjugated carbonyl groups. Therefore enzyme-catalyzed monoreduction of 251 is anticipated to be readily achieved by microorganisms. However, two problems interfered with attempts to obtain a practical asymmetric bioorganic synthesis of enantiomerically pure 207 from the vinylogous α-diketone 251. First, reduction of the C=C bond often accompanied C=O reduction. Saturation of α,β-unsaturated ketones is a common microbiological transformation. This undesired side reaction was prevented by microbiological reduction of 251 in the presence of excess 2-cyclohexenone or methyl vinyl ketone as competitive substrates for the C=C bond reductases but not for the C=O reductases.16 Furthermore, with a wide variety of microorganisms, reduction of the incipient 9-keto group generating 256 competed with the desired reduction to generate 207 with a hydroxyl at the incipient 11-position.
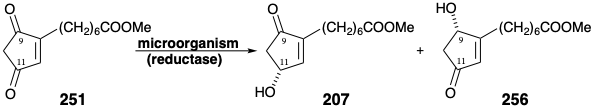
A less direct alternative strategy for enantioselective synthesis of 207 by asymmetric carbonyl reduction exploits functionality and unsaturation level adjustment of a hydroxydione precursor 257 that might be available by enantioselective (generating a pure enantiomeric product from an achiral precursor) reduction of a trione 258. Polar analysis of 258 suggests a synthesis of this dissonant functional array by the condensation of a methyl ketone 259 with the dissonant diester, diethyl oxalate.
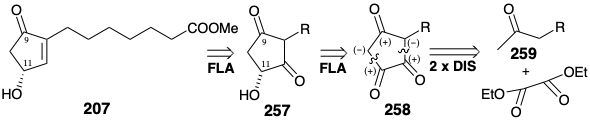
The trione 258 actually exists as an enol 258e that is a hydroxy derivative of 251. But this enol can be expected to have less proclivity than 251 toward saturation because the hydroxyl substituent will reduce the electrophilicity of the C=C bond by donation of a nonbonding electron pair. Furthermore, the carbonyl group at the incipient 11-position in 258e should be especially susceptible to nucleophilic attack by hydride owing to the dipole effect of a vicinal hydroxyl group. A synthesis of trione 258 began with azelaic acid (260) and its dimethyl ester to afford monomethyl azelate (261) by thermal equilibration. Condensation of the derived imidazolide with the magnesium enolate of lithium monomethyl malonate followed by decarboxylation, hydrolysis, and a second decarboxylation delivered methyl ketone 262. Claisen condensation with dimethyl oxalate and subsequent Dieckmann cyclization and methylation of the carboxyl group produced the key intermediate 258.
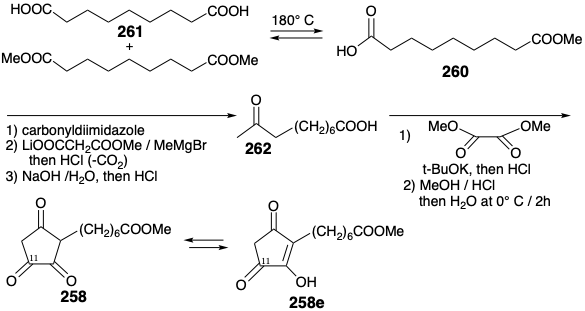
Trione 258 was cleanly and regioselectively reduced to hydroxydione 263 by a variety of microorganisms. Dipodascus uninucleatus catlayzed the completely asymmetric reduction of 258 to the 11(R) alcohol that is required for the total synthesis of prostaglandins. In contrast, Mucor rammanianus reduced 258 to the 11(S) alcohol. Conversion of optically pure hydroxydione 263 into the optically pure hydroxycyclopentenone 207 required selective reduction of the carbonyl at position 12. This was achieved by selectively masking the carbonyl at position 9 as an enol mesitylenesulfonate 264 followed by hydride reduction (see section 3.7). Subsequent hydrolytic allylic rearrangement of the intermediate allylic alcohol 265 delivered 207.16
Enantiocontrol by Exploiting Chiral Nonracemic Starting Materials
Instead of using the asymmetry of natural products, e.g. enzymes, to induce asymmetry during conversion of prochiral precursors into chiral synthetic intermediates, the asymmetry of readily available natural products, e.g. sugars, can be incorporated into synthetic targets by conversion into chiral nonracemic intermediates for total syntheses. For 207, the chiral center at position 11 might be derived from a chiral center in a sugar. Since every carbon in a sugar is oxygenated, polar disconnection of chiral nonracemic 207 with the boundary condition of uncovering a sugar-derived chiral segment suggests a trihydroxy precursor 266 (see section 3.7). Note that 266 is a nucleophilic umpoled synthon generated by polar reactivity inversion (PRI) at the incipient 9-position carbonyl. Further polar disconnection suggests an α-carbomethoxy- stabilized nucleophile 267 and D-glyceraldehyde as electrophile. D-Glyceraldehyde should be available by oxidative cleavage of any D-sugar. An especially efficient synthesis is suggested by a dislocation involving reductive coupling to connect two molecules of glyceraldehyde. The axially symmetrical precursor D-mannitol is available by reduction of D-mannose.
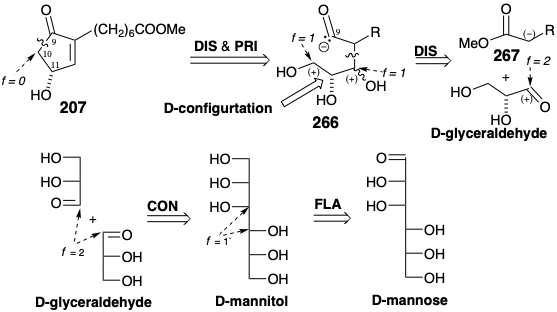
In an enantiospecific synthesis of optically pure 207 from D-mannitol, methyl oleate provided the nucleophile corresponding to 267.21 The C=C bond in methyl oleate comprises a latent carboxyl that is not unmasked until the end of the synthesis. Aldol condensation of methyl oleate with acetonide 269 of D-glyceraldehyde delivers 270. Deketalization unmasks a vicinal dihydroxyl array (see section 3.7) and subsequent lactonization differentiates the primary and secondary hydroxyls exploiting an internal masking group and the favorable stability of a temporarily bridged butyrolactone. The free primary hydroxyl in 271 is then activated by tosylation, and the α-ethoxyethyl (EE) ether of a cyanohydrin is generated from the lactone carbonyl by reduction, cyanohydrin formation, and O-alkylation with ethyl vinyl ether. Cyclization of 272 then completes the carbon skeleton. Removal of protecting groups, oxidative cleavage of the side chain C=C bond and methylation of the resulting carboxylic acid, hydrolysis of the cyanohydrin, and dehydration then delivers the optically pure hydroxy-cyclopentenone 207 from 273.
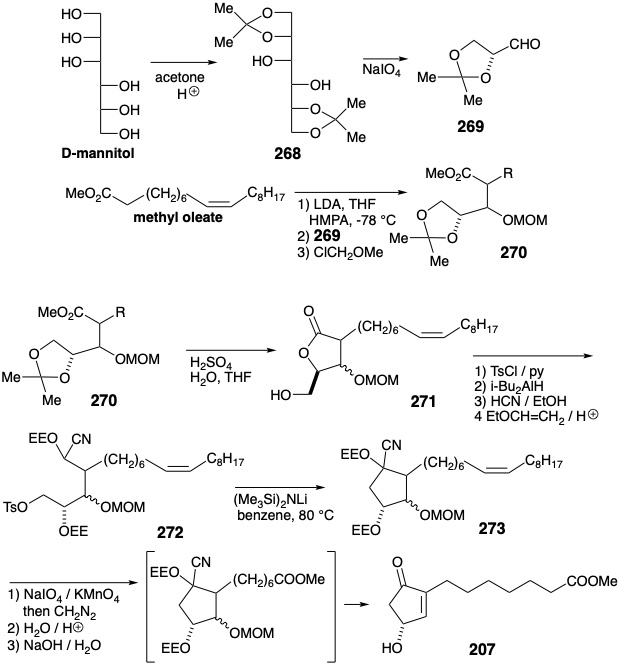
The abundant functional (oxygen on every carbon) and stereochemical information present in sugars suggests an even more ambitious synthesis of prostaglandins: incorporation of several sugar-related centers of chirality from a sugar starting material with a sugar-related hydroxyl at the incipient 10 position of PGF2α. Thus, polar disconnection of PGF2α with the boundary condition of uncovering a sugar-derived homochiral segment suggests a precursor 274 in which the target related polar reactivity implied by the hydroxyl at the 9-position in PGF2α must be inverted (PRI), e.g., as a nitrile-stabilized carbanion derived from an aldehyde cyanohydrin. Generalized representations 275 of 274, especially the Fischer projection 275c emphasize structural similarities with D-sugar precursors.
The necessity of deprotonating a cyanohydrin ether to generate 274 suggests replacement of the terminal carboxyl with a less reactive latent equivalent functional group such as an ether in 276 that also incorporates an ester as precursor for the cyanohydrin at the incipient 9-position with a view toward further polar disconnection of 276 to 278 is suggested by polar analysis which also reveals the possibility of disconnecting 278 to a methyl acetate nucleophile, or a malonic ester carbanion in which the added ester group serves as a reactivity control element (CEA), and a sugar derived electrophile 279. The required stereochemistry at the incipient 12-position in 278 would be generated by stereospecific inversion during the nucleophilic substitution of an oxygen electrofuge by a carbanion. This strategy may be derailed by an alternative possible SN2' nucleophilic substitution of the allylic nucleofuge in 2 7 9 . An alternative dislocation of subtarget 278 avoids this ambiguity. The sigma bond between incipient carbons 8 and 12 in 278 might replace a sigma bond between the incipient ester carbonyl oxygen and carbon 14 by a process involving allylic rearrangement of two π-bonds and a σ-bond in a precursor 280 by a cyclic three electron pair-shift. Such bond reorganizations, known generally as a [3.3] sigmatropic rearrangements, involve a temporary bridge in the transition state that, for the 280 to 278, conversion might be expected to adopt a chair-like conformation (SEC) as in 281. The rearrangement consequently involves predictable transfer of chirality from the migration origin at position 14 in 280 to the migration terminus at position 12 in 278. The driving force for sigmatropic rearrangements is a net increase in thermodynamic stability.
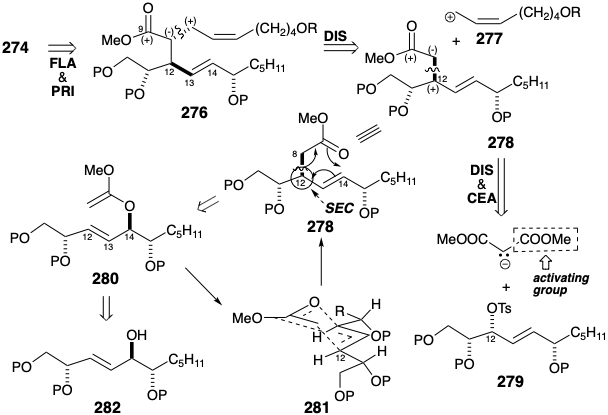
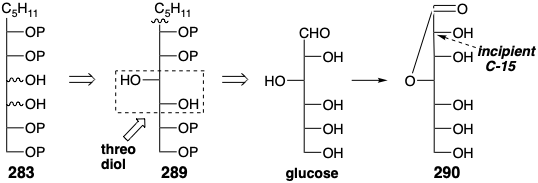
For the 280 to 278 conversion, an enol ester-Claisen rearrangement, this energetic advantage accrues from the generation of a C=O bond at the expense of a C=C bond. The ketene acetal 280 is a derivative of the allylic alcohol 282. A sugar-like progenitor 283 for 282 is suggested by 1,2-dioxidative addition. Such an intermediate might, for example, be produced by nucleophilic addition of an n-pentyl nucleophile to D-glucose.

The conversion of diol 283 into trans alkene 282 must surmount several hurdles. Since the vicinal diol is surrounded by hydroxyl groups or derivatives of hydroxyl groups as in 284, reductive cleavage generating an intermediate or transition state resembling carbanion 285 might lead to β-elimination of the wrong vicinal oxyanion producing 287 rather than 286.
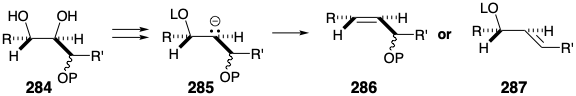
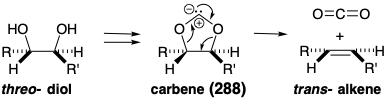
A more certain outcome can be assured by employing an alternative reaction, a concerted cycloelimination of carbon dioxide from a carbene-bridged derivative 288, to generate the requisite trans alkene from a vicinal diol. Such a process involves the cyclic shift of three electron pairs -- two σ-bonds and a nonbonding electron pair on the carbene carbon -- and is driven by the creation of two C=O bonds. Since the cycloelimination is concerted, the carbene derivative generated from a threo diol necessairly fragments to a trans alkene while that derived from an erythro diol would fragment to give a cis alkene. Therefore, to be a precursor of a trans alkene, the intermediate 283 must incorporate a threo diol as in 289 and glucose.
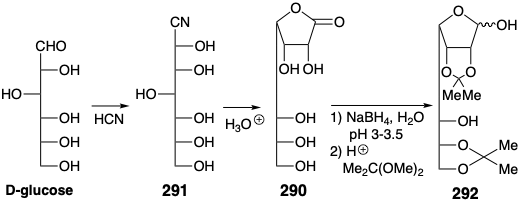
However, generation of 289 need not necessairly proceed directly from glucose by addition of an n-pentyl anion. In fact D-glycero-D-guloheptose (290), that can be prepared from glucose and incorporates the requisite configuration at the incipient position 15, is commercially available.
An enantiospecific synthesis of chiral nonracemic PGF2α was executed that derives three centers of chirality from glucose.22 Thus, glucose is chain extended by one carbon by addition of \(\ce{HCN}\). Acid-catalyzed hydrolysis and lactonization of the intermediate cyanohydrin 291 produces D-glycero-D-guloheptono-1,4-lactone (290). The hydroxyl groups in this intermediate must be differentiated to allow selective manipulation. Four hydroxyl groups can be masked by ketalization with acetone. Subsequent partial reduction of the lactone group delivers a lactol 292 that affords 293 upon further reduction and selective acetylation of a primary hydroxyl in the presence of two secondary hydroxyls. The threo diol array which remains unmasked in 293 may now be stereospecifically eliminated to generate a trans alkene.
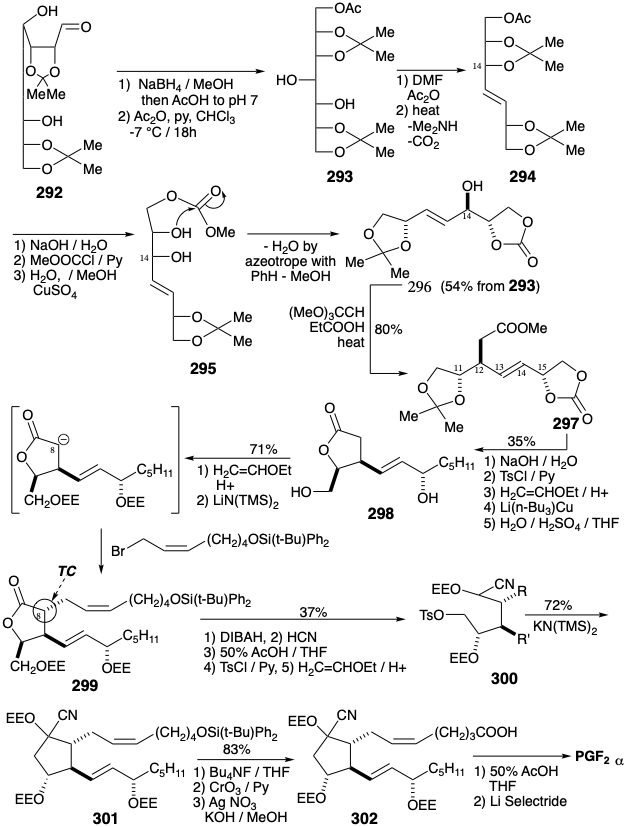
The requisite bridged carbene intermediate is generated by thermolysis of a dimethylformamide cyclic acetal derived from 293 in the presence of acetic anhydride. A concerted cycloelimination of this carbene then stereospecifically delivers the requisite trans alkene 294 in about 40% yield overall from 290. The masked allylic hydroxyl at the incipient 14-position must now be selectively unmasked to set the stage for an ortho ester-Claisen rearrangement. But both this hydroxyl and its vicinal neighbor are masked in 294 by the same acetonide. To selectively capture its neighbor after removal of the acetonide, the acetate in 294 was initially converted into a methyl carbonate that then intramolecularly acylates the neighboring hydroxyl in 295. One of two hydroxyls is thus protected by the temporary bridge of a carbonate. The remaining hydroxyl in 296 is then displaced with allylic rearrangement by a carbomethoxymethyl group. Thus, an orthoester Claisen reaction of 296 stereospecifically transfers the chirality of the hydroxyl substituted position 14 in 296 to a carbon substituted position 12 in 297. This intermediate incorporates three of the five chiral centers as well as the 13,14-trans C=C bond of the target PGF2α. Appendage of the carboxylic side chain, after masking the hydroxyls as α-ethoxyethyl (EE) ethers, was achieved by allylation of an ester enolate delivering 299. The stereochemistry at the incipient 8-position in 299 is a consequence thermodynamic control (TC) that favors a trans relationship between vicinal substituents on the five-membered lactone ring. Annulation of the cyclopentane ring required partial reduction of the lactone carbonyl, cyanohydrin formation, removal of the EE protecting groups, selective monotosylation of the primary hydroxyl and protection of the resulting triol as EE ethers. Finally treatment of 300 with base generated the corresponding nitrile stabilized carbanion which underwent intramolecular alkylation affording 301. A carboxyl was then generated after removal of the silyl protecting group. Removal of the EE protecting groups from the carboxylic acid 302 delivered a cyanohydrin that was cleaved to the corresponding ketone and reduced stereoselectively in situ to produce PGF2α. This efficient interception of the ketone carbonyl is a noteworthy tactic. The carbonyl was reduced in order to avoid loss of the hydroxyl at position 11 by dehydration of the base sensitive PGE2 intermediate.