
1.4: Polar Reactivity Analysis
- Page ID
- 285431

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)The π-electron pair in a C=C bond may be unevenly distributed if the π-bond bears or is conjugated with a functional group. It is reasonable to approximate the electron pair distribution in, for example, the enol or enolate π-bond as having electron abundance on the β-carbon since a hydroxyl substituent activates the β-carbon toward bond formation with a carbon electrophile. Thus, functional groups stabilize neighboring centers of electron abundance (nucleophilic centers) and centers of electron deficiency (electrophilic centers). For example, the carbonyl carbon of a ketone is electrophilic while the carbon α- to a carbonyl is potentially nucleophilic. Actual nucleophilicity at this carbon is obtained when this carbon is conjugated with the carbonyl carbon as the corresponding enol or enolate. Similarly, the enone C-C π-bond is electrophilic at the β-carbon while the γ-carbon is potentially nucleophilic, actual nucleophilicity being available by conversion to the corresponding enol or enolate. Thus, the polar activation afforded by a functional group may be extended to remote carbon centers by conjugation. This possibility is indicated for 23. In the ensuing discussion, centers of actual or potential electrophilic or nucleophilic reactivity will be designated as (+) and (-) respectively in contradistinction to centers of positive or negative charge, which will be designated as \(\oplus\) and \(\ominus\).
Actual nucleophilicity at this carbon is obtained when this carbon is conjugated with the carbonyl carbon as the corresponding enol or enolate. Similarly, the enone C-C π-bond is electrophilic at the β-carbon while the γ-carbon is potentially nucleophilic, actual nucleophilicity being available by conversion to the corresponding enol or enolate. Thus, the polar activation afforded by a functional group may be extended to remote carbon centers by conjugation. This possibility is indicated for 23. In the ensuing discussion, centers of actual or potential electrophilic or nucleophilic reactivity will be designated as (+) and (-) respectively in contradistinction to centers of positive or negative charge, which will be designated as \(\oplus\) and \(\ominus\).
Let us consider all possible synthetic strategies for direct synthesis of the butyryl moiety of butyrophenone (23) from two fragments by a polar reaction exploiting the polar activation afforded by the carbonyl group or any functional group precursor which confers electrophilicity to the same carbon atom. There are three possible C-C disconnections of the target 23. The three possible C-C connective strategies are summarized in equations 9-11. The strategies can be considered first in general terms by representing the required reactive polar precursors as synthons. The carbonyl group in 23 provides electrophilic reactivity at carbon 1 allowing a synthesis by polar creation of the 1- 2 bond by reaction with a three carbon nucleophile. The carbonyl group in 23 also potentially provides nucleophilic reactivity at carbon 2 allowing a synthesis by polar creation of the 2-3 bond by reaction with a two carbon electrophile. The carbonyl group in 23 also potentially provides electrophilic reactivity at carbon 3 allowing synthesis by polar creation of the 3-4 bond by reaction with a one carbon nucleophile. Usually the requisite synthons are recognized but only appropriate synthetic equivalents of these synthons are considered explicitly. For each of the above syntheses a large variety of alternative synthetic equivalents are possible. It is only necessary that the electrophilic precursor chosen has a functionality level one unit higher than the corresponding carbon in the target ( or has a C=C bond conjugated with an electrophilically activating functional group) and that the nucleophilic precursor has a functionality level one unit lower than the corresponding carbon in the target in order to achieve a direct synthesis of the target.

An indirect synthesis of the target may also be reasonable. For example, 23 could be prepared by addition of n-PrLi to benzaldehyde producing an intermediate 24 with the carbon skeleton of the target 23. Subsequent adjustment of functionality level by oxidation then delivers the ketone 23 (equation 12). Retrosynthetically such a strategy requires recognition of the possibility that benzaldehyde is a readily available electrophilic precursor of the benzoyl portion of 23. However, since the functionality level of this electrophile is the same at the incipient carbon 1 as in the target, the latter cannot be produced directly from benzaldehyde in a polar C-C bond forming process. Rather, the first dislocation of the target must be adjustment of its functionality level (FLA) prior to polar disconnection in the second dislocation (equation 13).
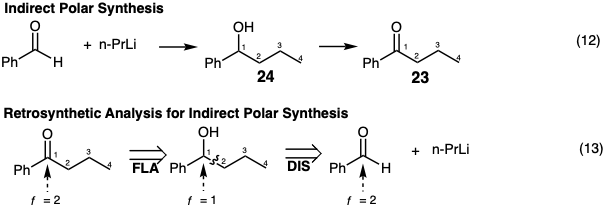
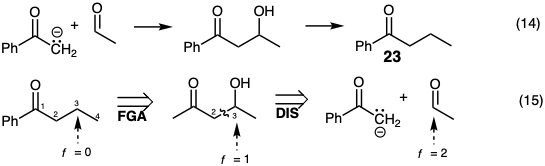
Topologically, the strategy of equation 13 is related to that of equation 9. An indirect synthesis of 23 by a strategy related topologically to that of equation 10 is outlined in equation 14. Retrosynthetically such a strategy requires recognition of the possibility that acetaldehyde is a readily available two carbon electrophile. However, since the functionality level of this electrophile is two units higher than the incipient carbon 3 in 23, a polar bond-forming reaction with a carbon nucleophile leads to a product in which the functionality level at this carbon is one unit higher than required for 23. Therefore, the first dislocation of the target must be adjustment of its functionality level (here functional group addition, FGA, a special case of FLA) prior to polar disconnection in the second dislocation (equation 15).
Regioselective Polar Reactions
It is important to recognize that all of the strategies considered above are hypothetical. The desired synthetic reaction between the chosen intermediates may not be the only reaction pathway available. For example, delocalized synthons are inherently ambident; they possess several centers of reactivity. Thus, the enolate in equation 14 is an ambident nucleophile that may react with an electrophile either at the carbonyl oxygen or α-carbon. Similarly, the conjugated electrophile 25 may react with a nucleophile either at the carbonyl carbon (1,2-addition) or the β-carbon (1,4- or Michael addition). Thus, 25 is an ambident electrophile. To be synthetically useful, bond formation must be accomplished at the desired position of an ambident nucleophile or electrophile, the reaction must be regioselective.

Reactivity Control Elements
We have now seen that indirect strategies, that involve dislocations of a target that do not directly reduce molecular complexity, may be desireable because: (1) they allow a subsequent dislocation of the target that efficiently simplifies molecular complexity, (2) they expliot readily available starting materials that have a high level of molecular complexity, or (3) they expliot certain readily available electrophiles or nucleophiles that have functionality levels that are inappropriate for direct C-C connective synthesis of a target. Indirect strategies may be desirable for other reasons. Thus, some atoms or groups of atoms may be exploited to control synthetic reactions by altering selectivity. We will refer to such molecular fragments as control elements.
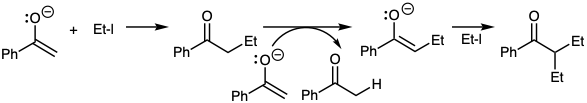
For example, alkylation of ketones as in equation 10 often results in polyalkylation owing to rapid proton transfers from product ketone to starting enolate. The strong basicity of ketone enolates also can result in proton abstraction from alkyl halides (β-elimination) rather than nucleophilic substitution. The addition of a carboethoxy group provides a less reactive less basic nucleophile that can be alkylated in good yield (equation 16). Such a carboethoxyl group is often referred to as an activating group since it activates the molecule toward proton abstraction. It is perhaps more significant, however, that this group deactivates the resulting nucleophile making it a more selective reactant. After it has served its purpose, the control element must be removed. Retrosynthetically, the desirability of exploiting a control element requires addition of that element (CEA) in the first dislocation of the target prior to the reaction it is intended to control which then is the second dislocation of the target (equation 17).

Difunctional Targets
Polar syntheses of difunctional targets by strategies that exploit the polar activation provided by both functional groups to achieve C-C bond formation may be divided into two categories: those whose carbon skeleton can be assembled directly with the required functionality and those that cannot. C-C connective syntheses that directly generate difunctional targets with the required functionality are possible if both functional groups impart the same actual or potential polar reactivity to the atoms connecting the functional groups. We will refer to such functional groups and the atoms connecting them as consonant circuits. For example, 26 and 27 contain consonant circuits. Direct C-C connective synthesis of constant difunctional targets can be achieved by: (a) conjugate addition to an electrophile whose unsaturation level is one unit greater than that of the target, or (b) nucleophilic substitution or addition to an electrophile whose functionality level is one unit greater than that of the target. It should be noted that, while the carbon skeleton is assembled directly with the correct functionality level in the above strategies, it is not always possible to achieve a direct synthesis of consonant difunctional targets with the required unsaturation level. Thus, the reaction of 28 with 29 will generate a product with the carbon skeleton of 27 but with greater unsaturation. Thus, the 27 ⇒ 28 + 29 dislocation must be a two step process.

Indirect strategies are also possible for assembling consonant difunctional targets. Thus, during C-C bond formation, an intermediate may be generated that does not have the proper functionality level for the desired target. However, while the functionality levels of the precursors may be different than those of the target, the functionality in the synthetic equivalents of those precursors is electronically the same as the functionality of the consonant difunctional target. We shall refer to functionality in precursors whose nucleophilicity or electrophilicity is the same as that of the target -- but whose level may differ -- as target-related functionality.
Polar reactivity dissonance is present in difunctional targets if the polar reactivity imparted to the connecting atoms by one functional group is reversed by the other. We will refer to such functional groups and the atoms connecting them as dissonant circuits. For example, 30 contains a dissonant circuit. Syntheses of dissonant difunctional targets can never be achieved by C- C connective routes that directly exploit the polar activation afforded by both functional groups. For example, polar disconnection of 30 at the 2,3-bond must generate an acyl nucleophile synthon 32. But the carbonyl group usually provides electrophilic reactivity as in 31 and not nucleophilic reactivity at the carbonyl carbon. Synthetic equivalents such as 34 of such abnormal synthons, i.e. acyl carbanion equivalents, are known. They contain functionality that is related to that in the target but in which the usual polar reactivity of the target functionality is masked and the opposite polar reactivity is stabilized.

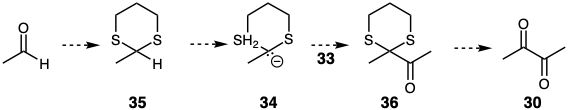
The normal polar reactivity of functional groups can be masked by conversion to unreactive derivatives.11 The functional group is said to be masked, blocked or protected in such derivatives The unreactive functional groups thus created are called masking or protecting groups. Such groups are examples of reactivity control elements. There is a subclass of masking groups which not only block the normal polar reactivity of a functional group but also facilitate the opposite polar reactivity. Such inversion of the polar reactivity of a functional group has been called umpölung.12 For example, the normal electrophilic reactivity of the carbonyl carbon in acetaldehyde can be transformed to nucleophilic reactivity by deprotonation of the derived thioacetal 35. The dithioacetal group not only masks the electrophilicity of the carbonyl precursor but also facilitates deprotonation of the carbonyl carbon by stabilizing the derived carbanion. Thus, the anion 34 is a synthetic equivalent of the acyl carbanion synthon 32. Acylation of 34 would deliver 36 from which the dissonant target 30 can be derived by hydrolysis. Note that the carbonyl group in the acetaldehyde starting material is exploited indirectly, i.e. after inversion of its usual polar reactivity, for the polar generation of a target C-C bond.
Polar disconnection of 37 at the 2,3-bond must generate a synthon 32 or 38 with inverted polar reactivity while disconnection at the 3-4 bond must generate a synthon 44 with inverted polar reactivity. It should be noted that synthetic equivalents of synthons with inverted polar reactivity, e.g. 34, 39, 41, and 45, by definition are dissonant molecules. Thus, for example, while the masked carbonyl carbon has nucleophilic reactivity in 41, the nucleofugacity of thiophenyl groups also makes this carbon potentially electrophilic. In this case the opposing polar reactivities are conferred by a single functional group, the dithioacetal, a functional group that can provide both nucleophilic and electrophilic activation at the same carbon.

An acyl carbanion equivalent 41 is available by deprotonation of the di(phenylthio)acetal derivative of acetaldehyde. Conjugate addition of carbanion 41 to methyl vinyl ketone might produce the thioketal 46 from which the dissonant target 37 would be obtained by hydrolysis.

A large variety of synthetic equivalents of "umpoled synthons" are available.13 Although they incorporate masked functionality with inverted reactivity, they are not necessarily prepared by umpolung of the target related functional group. For example, the synthetic equivalent 48 of the synthon 47 is an acetaldehyde enolonium ion equivalent. It reacts with cyclohexanone enolate to deliver 49 from which the dissonant target 50 is obtained upon hydrolysis.14

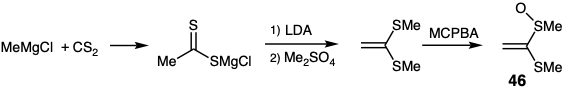
The acetaldehyde enolonium ion equivalent 48 can be obtained from methyl magnesium chloride and carbon disulfide by deprotonation of the intermediate dithioacetate followed by S-methylation and then by selective oxidation with m-chloroperbenzoic acid (MCPBA).14
Dissonant Targets from Dissonant Precursors
C-C connective polar syntheses of dissonant difunctional targets may also be achieved by multistep sequences employing polar reactions that exploit the polar reactivity provided by only one of the two functional groups in a dissonant precursor. For example, allylation of acetone enolate with 2-methoxyallyl bromide (51), a synthetic equivalent of the acetone enolonium synthon (43), followed by hydrolysis of the enol ether intermediate 52 could afford the dissonant difunctional target 37. Although 51 is prepared from acetone, the polar reactivity provided by the carbonyl group of the acetone precursor is not involved in the reaction of 51 with nucleophiles. Rather, the carbonyl group -- as an unreactive derivative -- is an innocent bystander. The polar reactivity required for C-C bond formation is provided by a target non-related second functional group, i.e. the bromo group. Also, it should be recognized that 51 is itself a dissonant difunctional molecule. The reaction of acetone enolate with 51 provides another example of a general principle: dissonant targets are available by polar C-C connective reactions of a dissonant precursor. Thus, 51 is a dissonant difunctional molecule.

As noted above for dithioacetals, some functional groups not only provide electrophilic reactivity at the carbon to which they are appended but also facilitate carbanion generation (i.e. reduction) at that carbon. Since this allows both electrophilic and nucleophilic reactivity at the functional carbon or any carbon conjugated with it, we shall refer to it as a biphilic functional group. For example, ≡N confers electrophilicity to carbon in a nitrile and also facilitates deprotonation of HC≡N to confer nucleophilicity to the same carbon. Thus, although the nitrile carbon in 53 is electrophilic and 53 is therefore a dissonant difunctional target, the cyanide ion is a stable nucleophilic equivalent of the nitrile carbanion synthon. The dissonant target 53 is available directly by the polar conjugate addition of cyanide to methyl vinyl ketone.

In summary, dissonant targets may be constructed by multistep sequences employing polar reactions that exploit: (a) inversion of the polar reactivity of one functional group in the target; (b) only one of the two functional groups in a dissonant precursor to provide polar reactivity; (c) a biphilic functional group.
Nonpolar Syntheses of Dissonant Targets
Dissonant difunctional targets are often prepared by direct C-C connective nonpolar reactions, such as oxidations, reductions, pericyclic bond-shift processes, or free radical additions. Thus, reductive coupling involves the union of two electrophilic centers accompanied by addition of an electron pair as in the pinacol reaction of acetone to produce the dissonant difunctional product pinacol.

Oxidative coupling involves the union of two nucleophilic centers accompanied by removal of one electron pair. The dissonant diketone 54 is obtained upon oxidative coupling of pinacolone enolate.

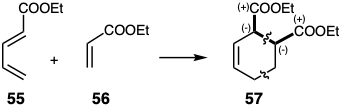
Generation of dissonant targets by pericyclic bond-shift processes is possible since the orientation of such reactions is controlled by p-orbital overlap which does not necessarily correspond with polar reactivity. For example, dissonant diester 57 is the major product from the cycloaddition of 55 with 56.
The free radical chain reaction between acetaldehyde and acetal 58 to generate 59 exemplifies another nonpolar C-C connective route to dissonant difunctional products.

Dissonant difunctional targets are also available by non-C-C connective processes such as addition of electronegative atoms X and Y to both carbons of a C=C bond. We shall refer to such reactions as dioxidative additions since both carbons are oxidized. Dioxidative additions can also occur with polyenes. Such reactions, which we shall call 1,n-dioxidative additions, always generate dissonant difunctionality. Thus, the conversion of 60 into 61 involves 1,2-dioxidative addition while the 62 to 63 conversion is a 1,4-dioxidative addition.

Disconnection of C=C Bonds
Retrosynthetic dislocations of a synthetic target involving disconnection of a carbon-carbon double bond, i.e. double disconnections, usually correspond to multistep synthetic sequences. There are no polar reactions that generate two bonds between two carbon atoms in a single step. (Note that dimerizations of carbenes are cycloadditions, that by definition, generate two bonds in a single step.) Therefore, if polar activation is to be exploited, a double connection during the synthesis must be made in two steps: the first, a polar union; the second, an elimination. The elimination step usually involves loss of HX, XY, or MX where X and Y are groups that are more electronegative than carbon while M is any group that is more electropositive than carbon. Thus retrosynthetically, disconnection across a C=C bond requires an addition as the first dislocation of the target.

The synthesis outlined in equation 18 is a representative example of the first approach. Here an electrophilic activating group of functionality level = 1 resides on each carbon. The syntheses outlined in equations 19-21 are representative examples of each of the last three strategies. In each case, the functionality level of the electrophilic synthon decreases by two on going to the C=C target. The functionality level of the nucleophilic synthon goes from -1, -2, or 0 for equations 19, 20, and 21 respectively in the precursors to 0 in the products. Also note that the second steps in equations 18 and 19 require oxidation of a hydrogen (deprotonation), and that the second step in equation 20 is a direductive elimination.

Strategic Flaws
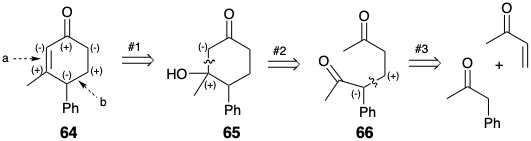
The polar activation provided by a single functional group in a target can be exploited numerous times during a synthesis to facilitate several C-C connective steps. For example, the carbonyl group of the 2-cyclohexen-1-one 64 might be used to provide the requisite nucleophilic and electrophilic reactivity for generating two C-C connections in this target (bonds a and b) corresponding to dislocations #2 and #3 (synthetic steps 1 and 2) in a strategy for synthesis of 64 from methyl vinyl ketone and phenyl acetone. The first dislocation of the target, addition of water to the C=C bond shows the necessary intervention of an intermediate during the double connection (synthetic steps 2 and 3) corresponding to the 65 to 64 conversion. This synthetic plan also provides an example of a significantly flawed synthetic design since the intermediate δ-diketone 66 cyclizes in two different ways only one of which affords the desired product. The isomeric 2-cyclohexen-1-one 67 can even be the major product of this reaction.15

Theory and Practice
Besides failures to achieve the required regioselectivity during addition reactions of multidentate nucleophiles or electrophiles, or the reactions of molecules with several similar functional groups, the planned removal of activating functionality, or unsaturation, as well as the introduction, removal, or interconversion of functionality by oxidation, reduction, metathesis, etc., may not be feasible owing to limitations of known reactions and/or limitations imposed by the reactivity characteristic of a particular synthetic target. Polar reactivity analysis serves merely to systematically generate a set of synthetic strategies. These must be subsequently evaluated in terms of the availability of suitably selective reactions and appropriate starting materials.