6.7: Lycopodine
- Page ID
- 285660

\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\( \newcommand{\dsum}{\displaystyle\sum\limits} \)
\( \newcommand{\dint}{\displaystyle\int\limits} \)
\( \newcommand{\dlim}{\displaystyle\lim\limits} \)
\( \newcommand{\id}{\mathrm{id}}\) \( \newcommand{\Span}{\mathrm{span}}\)
( \newcommand{\kernel}{\mathrm{null}\,}\) \( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\) \( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\) \( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\id}{\mathrm{id}}\)
\( \newcommand{\Span}{\mathrm{span}}\)
\( \newcommand{\kernel}{\mathrm{null}\,}\)
\( \newcommand{\range}{\mathrm{range}\,}\)
\( \newcommand{\RealPart}{\mathrm{Re}}\)
\( \newcommand{\ImaginaryPart}{\mathrm{Im}}\)
\( \newcommand{\Argument}{\mathrm{Arg}}\)
\( \newcommand{\norm}[1]{\| #1 \|}\)
\( \newcommand{\inner}[2]{\langle #1, #2 \rangle}\)
\( \newcommand{\Span}{\mathrm{span}}\) \( \newcommand{\AA}{\unicode[.8,0]{x212B}}\)
\( \newcommand{\vectorA}[1]{\vec{#1}} % arrow\)
\( \newcommand{\vectorAt}[1]{\vec{\text{#1}}} % arrow\)
\( \newcommand{\vectorB}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\( \newcommand{\vectorC}[1]{\textbf{#1}} \)
\( \newcommand{\vectorD}[1]{\overrightarrow{#1}} \)
\( \newcommand{\vectorDt}[1]{\overrightarrow{\text{#1}}} \)
\( \newcommand{\vectE}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash{\mathbf {#1}}}} \)
\( \newcommand{\vecs}[1]{\overset { \scriptstyle \rightharpoonup} {\mathbf{#1}} } \)
\(\newcommand{\longvect}{\overrightarrow}\)
\( \newcommand{\vecd}[1]{\overset{-\!-\!\rightharpoonup}{\vphantom{a}\smash {#1}}} \)
\(\newcommand{\avec}{\mathbf a}\) \(\newcommand{\bvec}{\mathbf b}\) \(\newcommand{\cvec}{\mathbf c}\) \(\newcommand{\dvec}{\mathbf d}\) \(\newcommand{\dtil}{\widetilde{\mathbf d}}\) \(\newcommand{\evec}{\mathbf e}\) \(\newcommand{\fvec}{\mathbf f}\) \(\newcommand{\nvec}{\mathbf n}\) \(\newcommand{\pvec}{\mathbf p}\) \(\newcommand{\qvec}{\mathbf q}\) \(\newcommand{\svec}{\mathbf s}\) \(\newcommand{\tvec}{\mathbf t}\) \(\newcommand{\uvec}{\mathbf u}\) \(\newcommand{\vvec}{\mathbf v}\) \(\newcommand{\wvec}{\mathbf w}\) \(\newcommand{\xvec}{\mathbf x}\) \(\newcommand{\yvec}{\mathbf y}\) \(\newcommand{\zvec}{\mathbf z}\) \(\newcommand{\rvec}{\mathbf r}\) \(\newcommand{\mvec}{\mathbf m}\) \(\newcommand{\zerovec}{\mathbf 0}\) \(\newcommand{\onevec}{\mathbf 1}\) \(\newcommand{\real}{\mathbb R}\) \(\newcommand{\twovec}[2]{\left[\begin{array}{r}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\ctwovec}[2]{\left[\begin{array}{c}#1 \\ #2 \end{array}\right]}\) \(\newcommand{\threevec}[3]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\cthreevec}[3]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \end{array}\right]}\) \(\newcommand{\fourvec}[4]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\cfourvec}[4]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \end{array}\right]}\) \(\newcommand{\fivevec}[5]{\left[\begin{array}{r}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\cfivevec}[5]{\left[\begin{array}{c}#1 \\ #2 \\ #3 \\ #4 \\ #5 \\ \end{array}\right]}\) \(\newcommand{\mattwo}[4]{\left[\begin{array}{rr}#1 \amp #2 \\ #3 \amp #4 \\ \end{array}\right]}\) \(\newcommand{\laspan}[1]{\text{Span}\{#1\}}\) \(\newcommand{\bcal}{\cal B}\) \(\newcommand{\ccal}{\cal C}\) \(\newcommand{\scal}{\cal S}\) \(\newcommand{\wcal}{\cal W}\) \(\newcommand{\ecal}{\cal E}\) \(\newcommand{\coords}[2]{\left\{#1\right\}_{#2}}\) \(\newcommand{\gray}[1]{\color{gray}{#1}}\) \(\newcommand{\lgray}[1]{\color{lightgray}{#1}}\) \(\newcommand{\rank}{\operatorname{rank}}\) \(\newcommand{\row}{\text{Row}}\) \(\newcommand{\col}{\text{Col}}\) \(\renewcommand{\row}{\text{Row}}\) \(\newcommand{\nul}{\text{Nul}}\) \(\newcommand{\var}{\text{Var}}\) \(\newcommand{\corr}{\text{corr}}\) \(\newcommand{\len}[1]{\left|#1\right|}\) \(\newcommand{\bbar}{\overline{\bvec}}\) \(\newcommand{\bhat}{\widehat{\bvec}}\) \(\newcommand{\bperp}{\bvec^\perp}\) \(\newcommand{\xhat}{\widehat{\xvec}}\) \(\newcommand{\vhat}{\widehat{\vvec}}\) \(\newcommand{\uhat}{\widehat{\uvec}}\) \(\newcommand{\what}{\widehat{\wvec}}\) \(\newcommand{\Sighat}{\widehat{\Sigma}}\) \(\newcommand{\lt}{<}\) \(\newcommand{\gt}{>}\) \(\newcommand{\amp}{&}\) \(\definecolor{fillinmathshade}{gray}{0.9}\)Biosynthesis of Alkaloids from L-Lysine
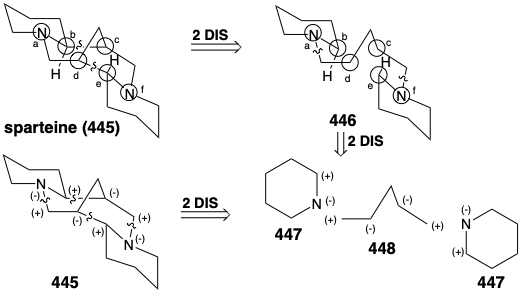
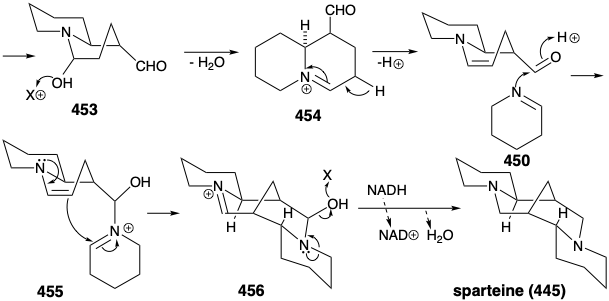
A variety of topolgically complex saturated nitrogen heterocycles are constructed in nature from simple acyclic precursors. The incisive logic of these biosyntheses is especially striking when viewed either from a topological or a polar reactivity standpoint. For example, the efficiency with which the intricate multicyclic skeleton of sparteine (445) is assembled, exclusively from three molecules of a symmetrical synthon, is remarkable. Topological analysis of 445 reveals the presence of six common atoms a-f. Cleavage of two bonds between two pairs of common atoms, b-c and d-e, simplifies the topology to two piperidine rings joined by a straight chain in 446. This intermediate is readily derived from two five carbon synthons 447 and 448. Polar reactivity analysis of 445 reveals that polar reactions, activated by the amino groups in 445 should readily allow its construction from 447 and 448.
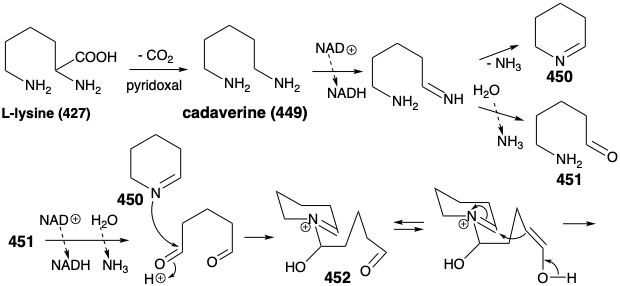
In fact, functionalized synthetic equivalents for both 447 and 448 are prepared in nature from L-lysine (427). Thus, pyridoxal catalyzed decarboxylation of 427 produces the symmetrical diamine 449. Oxidation and hydrolysis of 449 via 451 afford pentanedial, which provides iminium derivative 452 by reaction with 450. Intramolecular aldol condensation then affords 453. The iminium derivative 454 from dehydration of 453 yields an iminium derivative 455 by reaction of the corresponding enamine with a second equivalent of the imine 450. A second intramolecular aldol condensation affords 456. Dehydration and reduction provides sparteine (445).
Biosynthesis of Lycopodine
We have seen that many natural products are formed from a single starting material, such as (a) many polyketides, fatty acids, or prostaglandins from acetyl CoA, (b) many alkaloids from shikimic acid, or (c) terpenes from mevalonic acid. However, some natural products are formed by mixed biosyntheses from combinations of these starting materials. Thus, lysergic acid (see section 6.4) arises from chorismic acid plus the mevalonic acid-derived isopentenyl pyrophosphate plus a sugar, D-ribose. Similarly, indole alkaloids (see section 6.5) arise from chorismic acid plus a mevalonic acid-derived terpene, secologanin, plus a sugar, D-ribose.
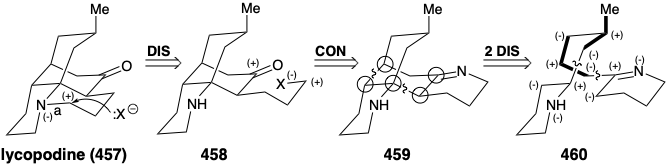
Now we shall see that the bridged multicyclic skeleton of the alkaloid lycopodine (457) arises from acetoacetyl CoA plus L-lysine (427). As for sparteine above, both topological and polar analysis of the biosynthetic strategy for 457 reveal its incisive logic. The carbonyl group in 457 is generated in nature by solvolytic cleavage of a temporary bridge. In the process, a propyl substituent with electrophilic activation at the end is generated, that is then used to construct the final ring of 457. Retrosynthetically, this involves disconnection to 458, followed by reconnection to 459. A considerable simplification of this subtarget results from disconnection of two bonds between pairs of common atoms in 459 to afford 460. Polar analysis of 460 reveals that reconnection of these bonds could be achieved by exploiting the polar activation provided by the nitrogen atoms in 460. Furthermore, 460 could be assembled from two large fragments by a polar reaction forming any of the bonds in the carbon chain connecting the two nitrogen heterocycles.
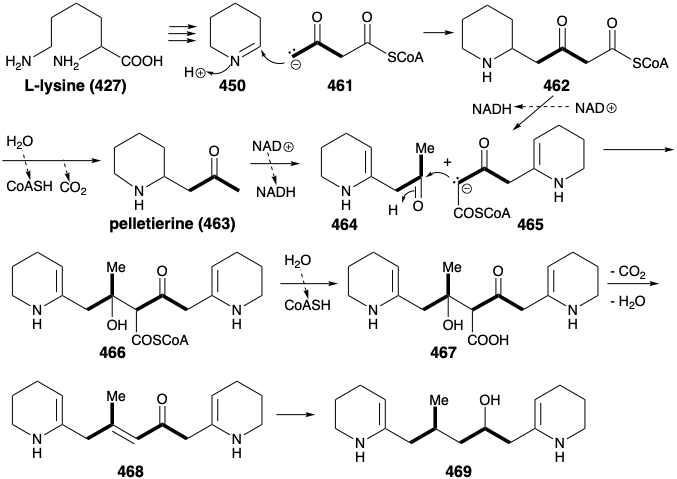
In nature, the two piperidine rings in 460 are derived from L-lysine (427), and the connecting chain is assembled from two three carbon units derived from actetoacetyl CoA (461). Aldol condensation of 461 with 450 affords 462. Note that alkylation of 461 occurs at the less acidic δ carbon. Perhaps this involves an enzyme-bound enamine derivative 470 (see below) of 461. Oxidation and deprotonation of 462 provides 465, while 462 also yields pelletierine (463) by hydrolysis and decarboxylation. Aldol condensation between 464 and 465 then provides 466, that is hydrolyzed to 467. Decarboxylative elimination gives 468, that is reduced to provide 469, a synthetic equivalent of the synthon 460 generated in the strategic analysis presented below.
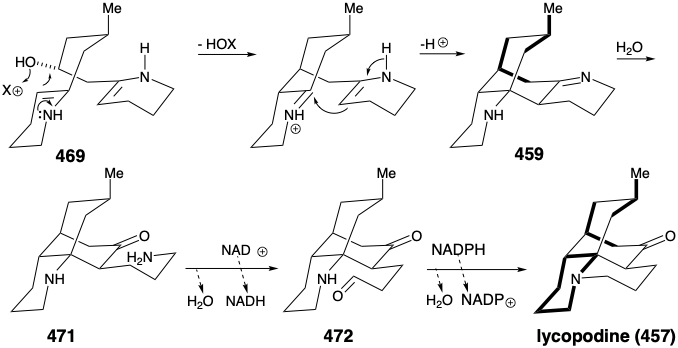
Cyclization of 469 by intramolecular enamine alkylation, followed by intramolecular aldol-like condensation, produces the intermediate 459 suggested in the strategic analysis. Hydrolysis of the imine in 459 generates the carbonyl group required for lycopodine. Oxidation of the resulting propyl amine 471 to an aldehyde 472 followed by intramolecular reductive alkylation then produces lycopodine (457) in which one lysine derived piperidine ring is clearly discernable while the two polyketide derived acetonyl units and a five carbon unit from a second molecule of lysine are intricately interwoven.
A Fatally Flawed Strategy for Lycopodine Synthesis
We have seen that both topological and polar analysis of the biosynthetic strategy for lycopodine (457) illuminate the logic of the process. The two functional groups in 457 can be exploited in a variety of strategies to facilitate construction of the skeletal network using polar reactions. There are five common atoms in 457, four carbon atoms that are all in ring B, and the nitrogen. We will first consider a fatally flawed strategy that only generates an epimer rather than the natural product itself.
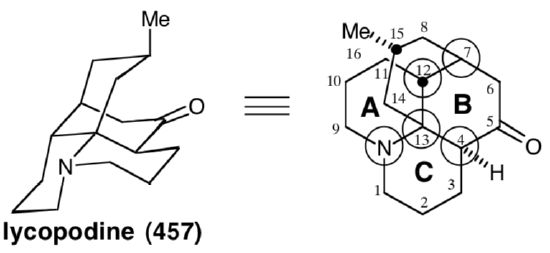
Polar analysis of ring B reveals that polar reactions, exploiting the polar activation afforded by the amino and carbonyl functionalities in 457, could be used to construct any bond of this ring. In the Wiesner approach to lycopodine,19 the final skeletal bond formed is between common atom 7 and noncommon atom 6, corresponding to the dislocation of 457 to 473. The penultimate bond formed is that between common atoms 4 and 13, corresponding to the dislocation of 473 to a bicyclic precursor 474. The elegance of the strategy lies in the plan to accomplish cyclization of the bicyclic intermediate 474 to the tetracyclic skeleton of the target 457 in a single step. The synthetic equivalent 475 of 474 has additional carbonyl groups at the 7 and 9 positions. The former provides additional electrophilic activation at C-7, while the latter deactivates the nucleophilicity of the amino group. The bicyclic intermediate 475 might be available from a symmetrical monocyclic precursor 476. The incipient amino group of 457, the nitrile nitrogen in 476, even provides polar activation for the construction of 476 from acrylonitrile and dihydroresocinal.
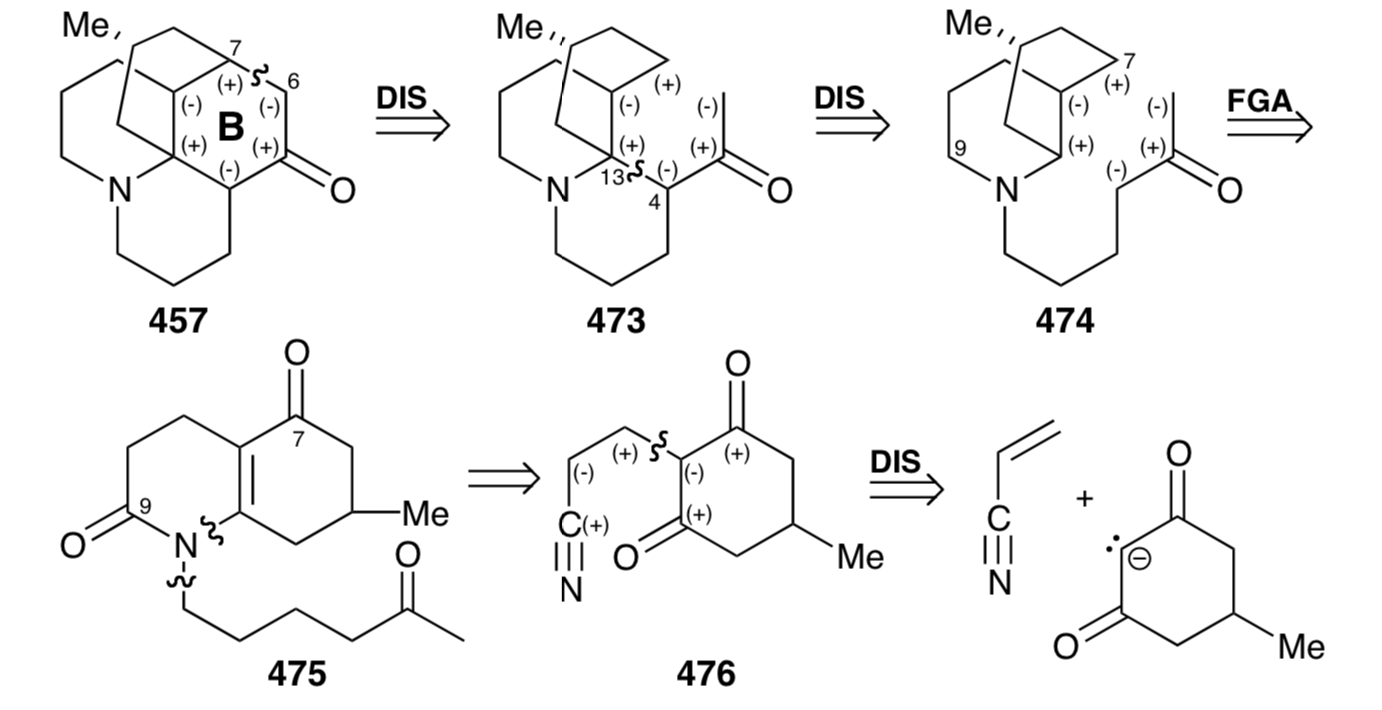
In fact, acid-catalyzed hydrolysis of 476 leads directly to the enamide 477 that afforded 475 by N- alkylation with the alkyl bromide 467 and subsequent hydrolytic removal of the masking ketal group. Base-catalyzed intramolecular Michael reaction of 475 could generate two stereoisomers at position 13 which result from addition of the carbanion to either face of the D-ring in 479. However, as expected, steric approach control fosters stereoselective addition on the side of the D-ring opposite the methyl substituent to afford an intermediate 481 rather than undesired stereoisomer 480. Nevertheless, the synthesis is fatally flawed because the subsequent aldol reaction gave exclusively 484, whose skeleton is epimeric with lycopodine at a C12. Thus, C-12 in the intermediate 481 is epimerizable, and the epimer 482 apparently cyclizes in complete preference to 481. This produces 484 rather than 483, that is required for the synthesis of lycopodine (457) . Reductive removal of the amide carbonyl and tertiary hydroxyl groups from 484 delivered 12-epi-lycopodine (485).

A Relay Strategy and a Symmetrical Precursor for Lycopodine
A second strategy for lycopodine synthesis generates ring D by cyclization of a tricyclic synthon 486 with preformed AB and C rings.20 This strategy was channeled by the prospect of exploiting a symmetrical fused tricyclic ketone 487 as a starting material. Thus, topological analysis of 457 recommends disconnection of two bonds between a common (circled) and a noncommon atom, the 7-8 and 13-14 bonds, to entirely remove the D-ring. A concomitant transposition of the carbonyl group from C-5 in 457 to C-6 is required to generate a symmetrical precursor 487. Furthermore, this transposition in 486 allows formation of the 7-8 C-C bond of the lycopodine skeleton by an intramolecular alkylation that exploits the polar activation afforded by a carbonyl at position 6. Whereas, the nitrogen in 486 can provide electrophilic activation for C-C bond formation at position 13 in 487. An amide carbonyl at C-9 in 486 is included as a deactivating group to decrease the nucleophilicity of the amino group disfavoring an undesired quaternization that might compete with alkylation of a carbanion nucleophile at C7.
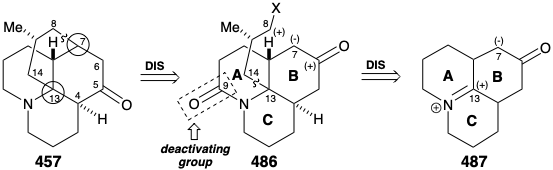
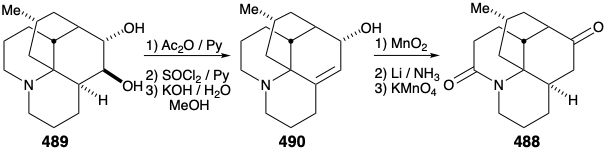
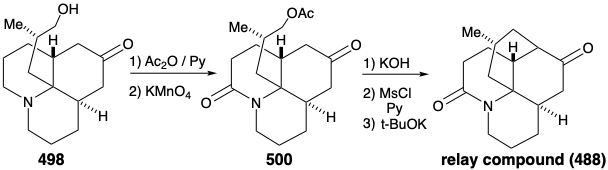
Intramolecular alkylation of the ketone 486 would yield 488. It would be reassuring if the final steps in the synthesis could be worked out with a sample of 488 that might be readily prepared from the natural product 457, perhaps via the diol 489, that had already been prepared from 457 during structural studies on the lycopodium alkaloids. This sample of compound 488 could then be used, instead of the synthetic material, to work out the details of the conversion of 488 to 457. This is another example of the strategem known as the relay approach, that we saw employed in syntheses of erythronolide B (see section 5.4) and quinine (see section 6.5). The advantage of this approach is that a valuable key intermediate can be obtained readily in quantity. The relay compound (e.g. 488) becomes the target of the synthesis.
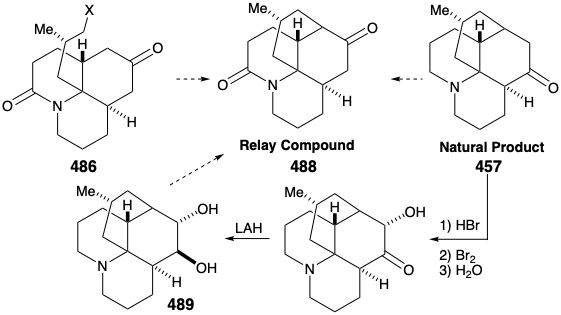
Let us first consider the interconversion of the relay compound 488 and 457 before examining the total synthesis of the relay compound. To differentiate the hydroxyls at positions 5 and 6, the diol 489 from natural lycopodine (457) was monoacetylated at the sterically most accessible C-6 hydroxyl. Dehydration followed by hydrolysis afforded 490. Oxidation of the allylic hydroxyl followed by reduction of the resulting α,β-unsaturated ketone and permanganate oxidation α to the tertiary amine gave the proposed relay compound, amide 488, in 13% overall yield from natural lycopodine (457).
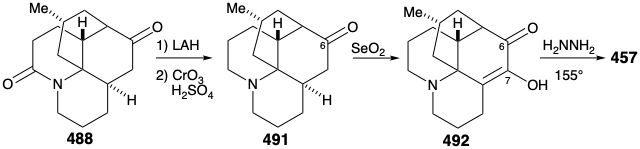
Reconversion of the relay compound 488 into lycopodine (457) was then achieved by removal of the amide carbonyl, by reduction with LAH, and oxidation of the resulting C-6 epimeric alcohols. The amino ketone 491 was then oxidized to the diosphenol 492, that was reduced selectively by a Wolff-Kishner reaction with hydrazine hydrate to afford lycopodine (457).
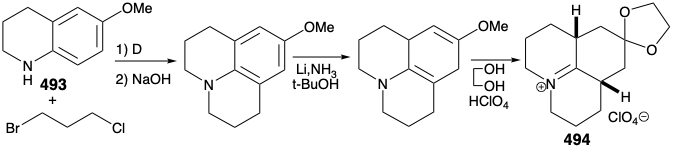
As noted above, a synthesis of 486, and hence the relay compound 488, from a symmetrical starting material was envisioned (see above). In particular, 486 might be prepared from 487 by reaction with a nucleophilic side chain synthon. With a provision for masking the electrophilicity of the carbonyl group, this strategy proved viable. Thus, 494 was prepared from thalline (493) by alkylation with 1-bromo-3-chloropropane followed by dissolving metal reduction and ketalization. Reaction of 494 with the nucleophilic fragment 495 followed by hydrolysis gives the cis,cis-fused tricyclic amine 496. Ring closure of this epimer is impossible. Epimerization to the trans,cis isomer 485 must precede ring closure. Therefore, epimerization was accomplished by exploiting the carbonyl functionality in 496 by bromination followed by Mattox-Kendall dehydrobromination and dissolving metal reduction. Demethylation of the product 497 then afforded a mixture of racemic diastereomeric ketones 498 and 499. These were separated by chromatography on alumina. The minor isomer 498 possessed the natural relative configuration of the methyl substituent. Thus, the present synthesis is nonstereospecific, and a near fatal major loss of valuable material occurs owing to the formation of an unwanted stereoisomer 499.
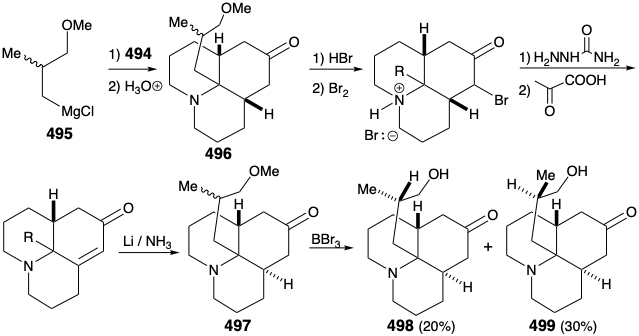
Before intramolecular alkylation of the tricyclic ketone could be accomplished, the nucleophilicity of the amino group had to be attenuated by conversion of the amino group in 498 into an amide 500 to avoid N-alkylation. Saponification, mesylation, and intramolecular alkylation then provided the relay compound 488 in racemic form. Since 476, albeit homochiral, derived from natural lycopodine (457) had already been converted to 457 as discussed above, the total synthesis was complete.
An Unintentionally Biomimetic Strategy
In the two strategies for lycopodine (457) discussed above, one generated ring B last and one generated ring D last. Now we shall consider a strategy that generates ring C last as found in the biosynthesis of 457. Moreover, in further analogy with the biosynthetic strategy, the three carbon chain substituent on ring B of 501, that is the used to complete ring C, is incorporated into a temporary ring in a precursor 502 by attachment at the (latent) carbonyl carbon at C-5. Finally, the consonant circuit between the amino and methoxy groups in the aromatic precursor 503 suggests a polar construction from 504 of the 4-13 C-C bond, again in analogy with the biosynthetic strategy, by attack of a C-13 electrophile on a nucleophilic center at the incipient C-4. However, this strategy was conceived before the biosynthesis of lycopodine had been elucidated. In the words of the author of the strategy, "although the particular synthetic plan followed for the construction of the tetracyclic system had no particular basis in biogenetic considerations, very recent work has suggested a biogenetic pathway in which the crucial cyclization step is strikingly similar to the one we devised."21
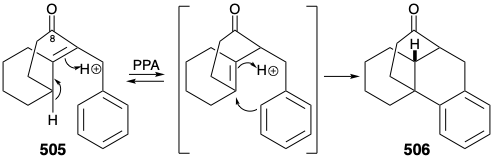
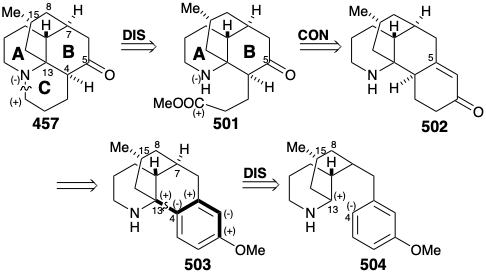
Before we consider the successful implementation of this plan, it is instructive to note that it was developed with the aid of lessons learned during attempts to achieve a synthesis using other, fatally flawed, strategies. One unsuccessful early plan for a transformation analogous to the generation of 503 from 504 was supported by successful model studies. Thus, a carbocyclic model 505 readily underwent an analogous cyclization to 506 on heating with polyphosphoric acid. It was anticipated that the methyl substituent at position 15 on the D-ring of 504 could be introduced subsequently by exploiting the nucleophilic activation afforded by a carbonyl group at position 8. Furthermore, the inclusion of this carbonyl group could enhance the general utility of the synthesis because oxygen substitution is found at position 8 in many lycopodium alkaloids. However, in contrast to the carbocyclic model 505, cyclization of the heterocyclic analogue 507 to give 508 could not be achieved. Rather 507 was apparently prone toward elimination leading to aromatization.
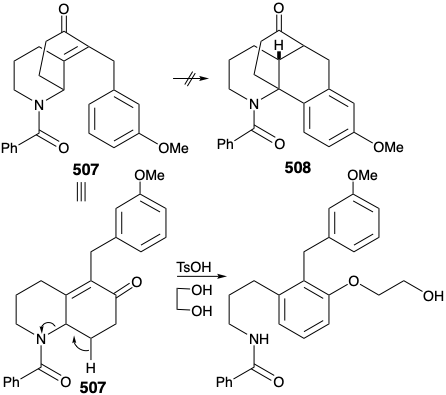
Nevertheless, an encouraging observation emerged from this model study. Thus, the dialkylated derivative 509, a byproduct in the synthesis of 507, underwent the desired type of cyclization. The success of this cyclization seemed attributable to two factors, an axial orientation of a benzyl substituent and blocking of the elimination pathway.
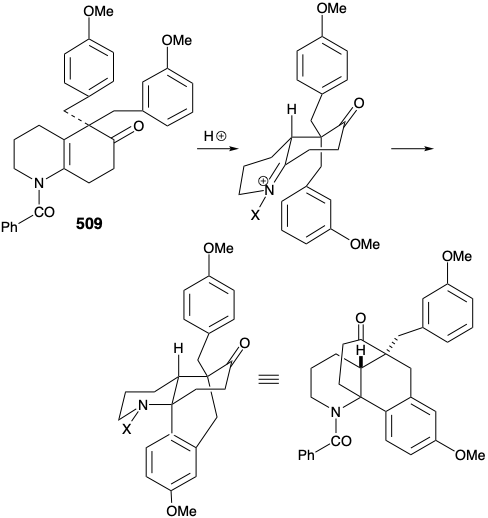
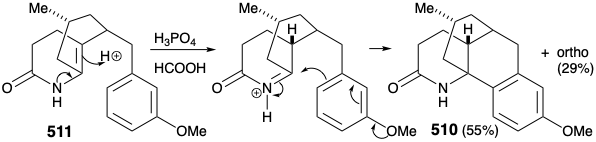
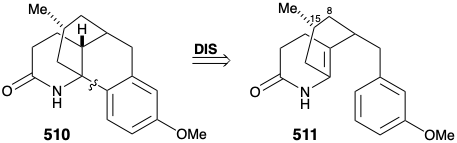
A modified strategy was then devised in which the methyl substituent required at position 15 was introduced prior to the annelation of ring B and the carbonyl group at position 8 was deleted. Moreover, the stereochemistry of this methyl substituent in lycopodine dictated a trans relationship of the methyl and m-methoxybenzyl groups in the new subtarget. The functionalized derivative chosen to embody these requirements was 511. It was further recognized that the trans methyl subtituent in 511 should virtually eliminate the energy barrier to achieving the axial orientation of the benzyl substituent required for cyclization to 510.
Several strategies were explored for synthesis of the subtarget 511, that is obviously derivable from 512. An approach to 512 via conjugate addition of a methyl nucleophile to 513 followed by oxidative cleavage of the cyclohexenone 514 was precluded by the proclivity of 514 to isomerize into the β,γ-unsaturated isomer 515.
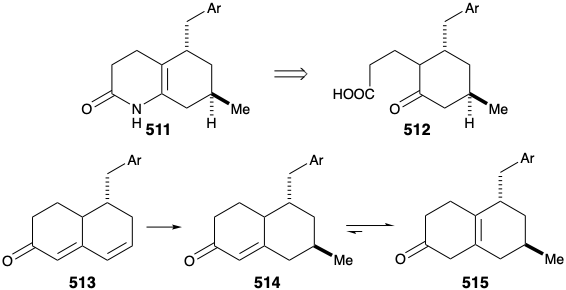
A second approach to 511 exploits the consonant circuit between the carbonyl carbon and enamide nitrogen in 511 or the related consonant circuit between the two carbonyl groups in keto amide 516. Disconnection of the three carbon side chain suggests a cyclohexanone precursor 517 that can be generated by polar connections activated by the carbonyl group.
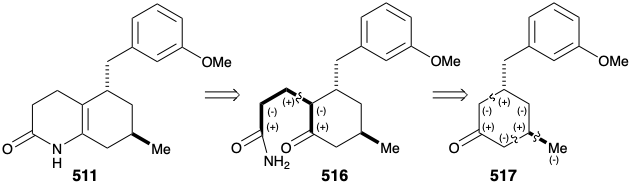
Cyclohexanone 517 was prepared by polar reactions between an acrylic ester and acetoacetic ester. Thus, a symmetrical dione 519 was generated from β,γ-unsaturated ester 518 by prototropic allylic rearrangement, followed by Michael addition of ethyl acetoacetate, Dieckmann cyclization, hydrolysis, and decarboxylation. Selective reduction of only one carbonyl group in 519 was facilitated by masking the second carbonyl of this dione as a vinylogous ester. Acid-catalyzed dehydration then provided a cyclohexenone that delivered 517 upon stereoselective 1,4-addition of a methyl nucleophile.
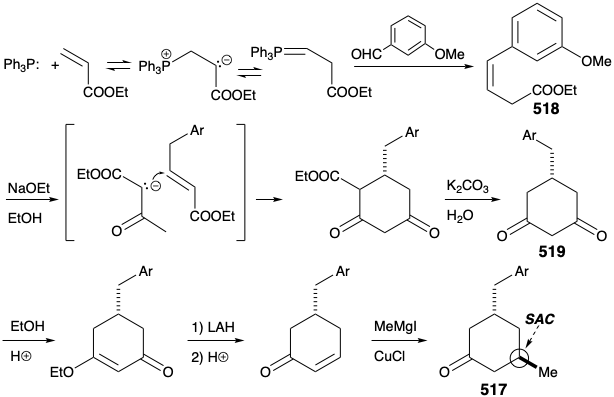
The original strategy for construction of key intermediate 511 from cyclohexanone 517 had one major shortcoming. Thus, conversion of 517 into 511 requires regioselective Michael alkylation. However, alkylation of 517 occurred nonregioselectively at both carbons α to the carbonyl producing a mixture of 511 and 520.
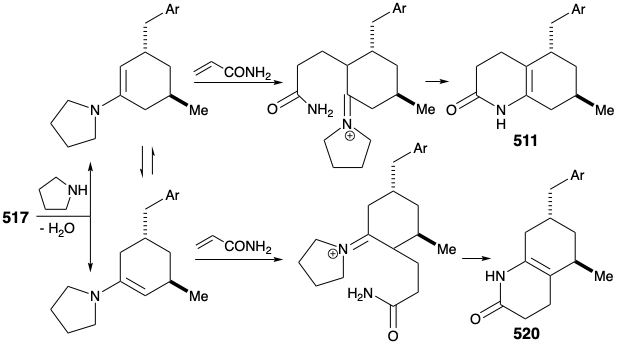
An improved, completely structurally specific synthesis of the subtarget 511 was finally devised that exploited regiospecific generation and electrophilic trapping of the enolate 522. This was achieved by Michael addition of a benzyl nucleophile to the cyclohexenone 521. The resulting regiospecific enolate was then alkylated with allyl bromide. The process is also highly stereoselective owing to a preference for axial attack in Michael additions of organocopper nucleophiles and a preference for an equatorial disposition of the methyl substituent in 521. Furthermore, the required trans relationship between the allyl and benzyl substituents is assured by thermodynamic control owing to epimerizability α to the ketone carbonyl. Hydroboration and oxidation of the allyl side chain, esterification, and reaction of the resulting ketoester with ammonia afforded the key intermediate 511.
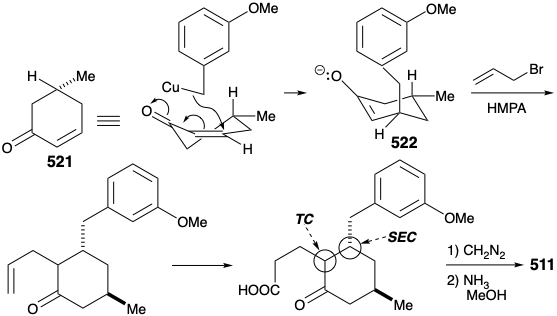
Intramolecular electrophilic aromatic substitution gave mainly the desired para substituted cyclization product 510 (55%) together with some ortho substitution product (29%). The amide carbonyl was then removed by reduction with LAH, and the protective aromaticity of the aryl ring was removed by Birch reduction. A plan to effect ring cleavage by oxidation of an α,β-unsaturated cyclohexenone failed owing to a thermodynamic preference for the required enone 502 to exist as the corresponding β,γ- unsaturated tautomer 524.
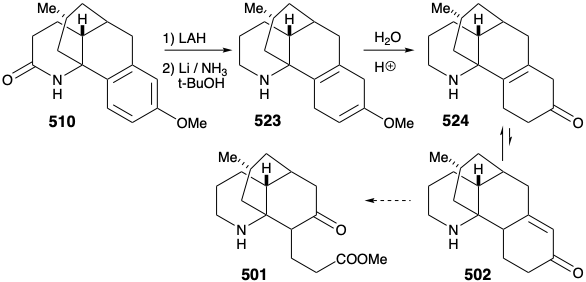
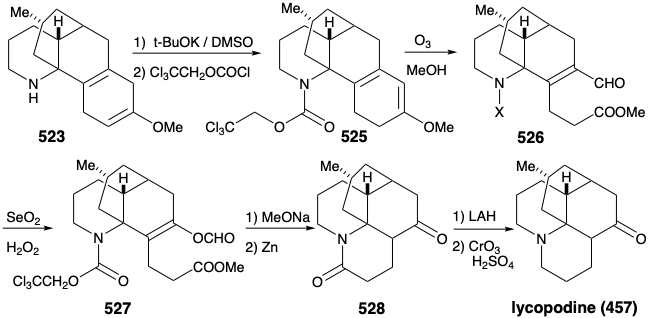
Once again, therefore, an alternative strategy had to be formulated. The original plan for generating 489 from 497 was fatally flawed. Indeed, while the each step in the original plan was well precedented, so was the likelihood that 502 would be in equilibrium with a substantial amount of 524. Indeed, this same problem derailed an attempted synthesis of 512 from 514 (see above). As is so often the case, a shortcoming of well-known methodology for achieving an important synthetic goal, especially if it impedes the conclusion of an ambitious total synthesis, inspires the application of novel chemistry to provide a solution to the dillema. Necessity is the mother of invention! Thus, generation of 501 from 523 required oxidative cleavage of two bonds, "a" and "b", in the cyclohexadiene ring. The original plan called for cleavage of bond "a" first after an isomerization that placed a readily cleavable C=C bond in this position. In the alternative strategy, bond "b" is cleaved first after an isomerization that placed a readily cleavable C=C bond in this position.
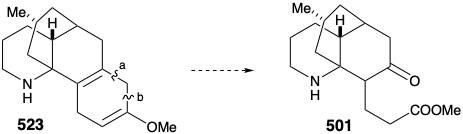
Thus, the 1,4-cyclohexadiene 523 was isomerized to a conjugated 1,3-diene, and the amino group was masked to protect it from oxidation. Selective ozonolysis of the more electron rich C=C bond in 525 then afforded the aldehydo methyl ester 526. An unusual Baeyer-Villager oxidation of 526 gave the enol formate 527 that afforded keto amide 528 after methanolysis of the enol ether, removal of the carbamate protecting group from the amino nitrogen, and lactamization. The amide carbonyl was then removed to provide lycopodine (457) by reduction with LAH followed by reoxidation of the C-5 hydroxyl to the required C-5 carbonyl group.